

Abstract
We have imaged, in real time, fluorescent tumors growing and metastasizing in live mice. The whole-body optical imaging system is external and noninvasive. It affords unprecedented continuous visual monitoring of malignant growth and spread within intact animals. We have established new human and rodent tumors that stably express very high levels of the Aequorea victoria green fluorescent protein (GFP) and transplanted these to appropriate animals. B16F0-GFP mouse melanoma cells were injected into the tail vein or portal vein of 6-week-old C57BL/6 and nude mice. Whole-body optical images showed metastatic lesions in the brain, liver, and bone of B16F0-GFP that were used for real time, quantitative measurement of tumor growth in each of these organs. The AC3488-GFP human colon cancer was surgically implanted orthotopically into nude mice. Whole-body optical images showed, in real time, growth of the primary colon tumor and its metastatic lesions in the liver and skeleton. Imaging was with either a trans-illuminated epifluorescence microscope or a fluorescence light box and thermoelectrically cooled color charge-coupled device camera. The depth to which metastasis and micrometastasis could be imaged depended on their size. A 60-μm diameter tumor was detectable at a depth of 0.5 mm whereas a 1,800-μm tumor could be visualized at 2.2-mm depth. The simple, noninvasive, and highly selective imaging of growing tumors, made possible by strong GFP fluorescence, enables the detailed imaging of tumor growth and metastasis formation. This should facilitate studies of modulators of cancer growth including inhibition by potential chemotherapeutic agents.
Keywords: cancer, animal model, fluorescence gene, external imaging
Current methods of external imaging of internally growing tumors include x-rays, MRI, and ultrasonography. Although these methods are well suited for the noninvasive imaging of large-scale structures in the human body (1), they have limitations in the investigation of internal growing tumors. In particular, monitoring growth and metastatic dissemination by these methods is impractical because they either use potentially harmful irradiation or require harsh contrast agents and, therefore, cannot be repeated on a frequent, real-time basis.
Optical imaging of cancers has been challenging because tumor cells usually do not have a specific optical quality that clearly distinguishes them from normal tissue. Also, conventional optical imaging has been severely limited by the strong absorbance and scattering of the illuminating light by tissue surrounding the target. As a result, neither the sensitivity nor spatial resolution of current methods is sufficient to image early-stage tumor growth or metastasis (2).
Previous attempts to endow tumors with specific, detectable spatial markers have met mostly with indifferent success. These included labeling with mAbs and other high-affinity vector molecules targeted against tumor-associated markers (3–7). However, results were limited due to a low tumor/background contrast and by the toxicity of the procedures.
Intravital videomicroscopy (IVVM) is another approach to optical imaging of tumor cells. IVVM allows direct observation of cancer cells but only if they are visible in the blood vessels (8). Even in this limited arena, IVVM does not lend itself to following tumor growth, progression, and internal metastasis in a live, intact animal.
A major conceptual advance in optical imaging was to make the target tumor the source of light. This renders the incident light scattering much less relevant. One early attempt inserted the luciferase gene into tumors so that they emit light (9). However, luciferase enzymes transferred to mammalian cells require the exogenous delivery of their luciferin substrate, an essentially impractical requirement in an intact animal. Also, it is not known whether luciferase genes can function stably over significant time periods in tumors and in the metastases derived from them.
A more practical approach to tumor luminance is to make the target tissue selectively fluorescent. Tumor-bearing animals were infused with protease-activated, near-infrared fluorescent probes (10). Tumors with appropriate proteases could activate the probes and be imaged externally. However, the system proved to have severe restrictions. The selectivity was limited because most normal tissues have significant protease activity. In fact, the normal activity in liver is so high as to preclude imaging in this most important of metastatic sites. The short lifetime of the fluorescence probes would appear to rule out growth and efficacy studies. The requirement of appropriate, tumor-specific protease activity and the requirement of effective tumor delivery of the probes also limit this approach (10).
We report here a new approach to producing tumors whose fluorescence can be viewed externally in intact animals. This is an extension of our previous work, which used stable green fluorescent protein (GFP) expression in cancer cells as an extremely effective tumor cell marker in conventional diagnostic dissections. The fluorescence-enhanced sensitivity illuminated tumor progression and allowed detection of metastases in exposed or isolated fresh visceral organs and tissues down to the single-cell level (11). Tracking of cancer cells that stably express GFP in vivo is far more sensitive and rapid than the traditional, cumbersome procedures of histopathological examination or immunohistochemistry. In particular, GFP labeling markedly improved the ability to visualize metastases in fresh soft organs and bone (11–18).
A major advantage of GFP-expressing tumor cells is that imaging requires no preparative procedures and, therefore, is uniquely suited for visualizing in live tissue (11–18). Using stable, high-GFP-expression tumor cells that we have selected (11–18), we demonstrate external, noninvasive, whole-body, real-time fluorescence optical imaging of internally growing tumors and metastases in transplanted animals.
Materials and Methods
Microscopy.
A Leica fluorescence stereo microscope (model LZ12) equipped with a mercury 50-W lamp power supply was used. Selective excitation of GFP was produced through a D425/60 band-pass filter and 470 DCXR dichroic mirror. Emitted fluorescence was collected through a long-pass filter (GG475; Chroma Technology, Brattleboro, VT) on a Hamamatsu C5810 three-chip cooled color charge-coupled-device camera (Hamamatsu Photonics Systems, Hamamatsu City, Japan). Images were processed for contrast and brightness and analyzed with the use of image pro plus 3.1 software (Media Cybernetics, Silver Springs, MD). High-resolution images of 1,024 × 724 pixels were captured directly on an IBM PC or continuously through video output on a high-resolution Sony VCR, model SLV-R1000 (Sony, Tokyo).
Doubling Time of Stable GFP Clones.
B16F0-GFP (18) or nontransduced cells were seeded at 1.5 × 104 in 35-mm culture dishes. The cells were harvested and counted every 24 hr with a hemocytometer (Reichert). The doubling time was calculated from the cell-growth curve over 6 days (data not shown).
Animals and Cell Injection.
Six-week-old female B57CL/6 mice were injected with 106 B16F0-GFP cells in the lateral tail vein. Cells first were harvested by trypsinization and washed three times with cold serum-free medium and then injected in a total volume of 0.2 ml by using a 1-ml 27G2 latex-free syringe (Becton Dickinson) within 30 min of harvesting. Six-week-old BALB/c nu/nu male and female mice were transplanted with 106 B16F0-GFP cells in the lateral tail vein or portal vein by using the same method as described above.
All animal studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health Guide for the Care and Use of Animals under assurance number A3873-1. Mice were fed with autoclaved laboratory rodent diet (Tecklad LM-485; Western Research Products, Orange, CA).
Surgical Orthotopic Implantation (SOI) (20).
Tumor fragments (1 mm3) from the liver-metastatic AC3488 tumor (21), stably expressing GFP after in vivo transduction (M.Y. and R.H.M., unpublished data), were implanted by SOI in nude mice. During proper exposure of the colon after a lower midline abdominal incision, the serosa of the colon was removed and two pieces of 1-mm3 tumor fragments per mouse were implanted. An 8-0 surgical suture was used to penetrate these small tumor pieces and suture them on the wall of the intestine, which then was returned to the abdominal cavity. The incision in the abdominal wall was closed with a 7-0 surgical suture in one layer (20). The animals were kept under isofluorane anesthesia during surgery. All procedures of the operation described above were performed with a ×7 magnification microscope (MZ6; Leica, Deerfield, IL). Animals were kept in a barrier facility under HEPA filtration (20).
Analysis of Metastases.
Periodically, the tumor-bearing mice were examined by whole-body fluorescence microscopy or in a fluorescence light box (Lightools Research, Encinitas, CA), as described above. In the case of C57BL/6 mice, hair was removed with Nair (Carter–Wallace, New York, NY).
Fluorescent Cross-Section.
A cross-section was made at the position, as shown in Fig. 5B, to simulate tomography and localize the external images. The animals were sacrificed and kept frozen after external fluorescence images were acquired. The whole mouse then was sliced in cross-section at approximately 1-mm thickness by using disposable microtome blades (Model 818; Leica). The sections then were observed directly under fluorescence microscopy.
Figure 5.
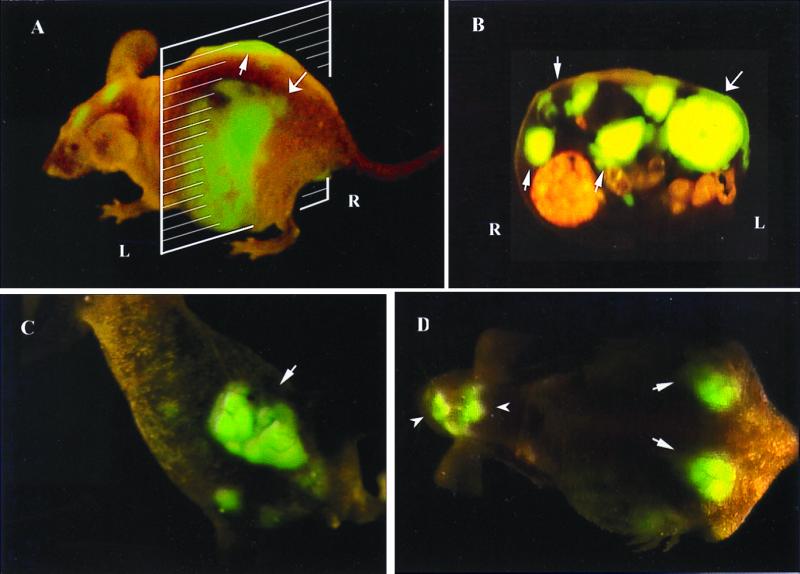
External and internal images of liver lesions of AC3488-GFP. (A) Lateral, whole-body image of metastatic liver lesions of a GFP-expressing human colon cancer in the left (thick arrow) and right lobes (fine arrow) of a live nude mouse at day 21 after surgical orthotopic transplantation. (B) Cross-section of mouse shown in A corresponding to the level of the external image of the tumor in the liver that was acquired (A). Fine arrows show metastatic lesions in the right lobe of liver, and the thick arrow shows the metastatic lesion in the left lobe of liver. (C) Fluorescent whole-body ventral image of primary colon tumor (arrow). (D) Dorsal external image of metastatic tumor in the caudal region of the left and right lobes of the liver (thick arrows) and skull metastasis (arrowheads).
Determination of Minimum GFP-Expressing Tumor Size Externally Imaged at Various Depths.
A Leica MZ12 fluorescence microscope coupled with a Hamamatsu C5810 three-chip cooled color charge-coupled-device camera was used to acquire the images as described above. The actual size of the GFP-expressing tumors or metastases externally imaged was measured directly by imaging the GFP-expressing tumor after direct exposure of the tumor-containing tissue. Tumor specimen size is expressed as a diameter assuming spherical geometry. The depth of the GFP-expressing tumors was determined in isolated tissues by acquiring calibrated images with image pro plus 3.1 software.
Results
Isolation of Stable, High-Level Expression GFP Transductants of B16F0-GFP Cells.
GFP- and neomycin-transduced B16F0 cells were selected previously in multiple steps for growth in levels of Geneticin (G418) up to 800 μg/ml and for high GFP expression (18). The selected B16F0-GFP cells have a strikingly bright GFP fluorescence that remains stable in the absence of selective agents after numerous passages (18) (Fig. 1). There was no difference in the doubling times of parental cells and selected transductants as determined by comparison of proliferation in monolayer culture (data not shown).
Figure 1.
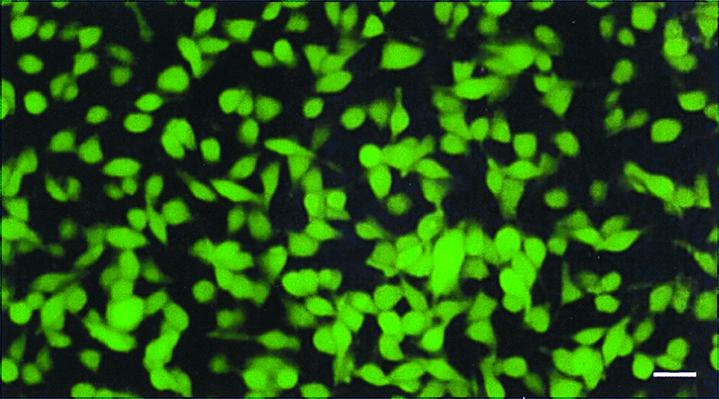
Stable, high-level GFP-expressing B16F0 murine melanoma transductants in vitro. The murine malignant melanoma cell line B16F0 was transduced previously with the RetroXpress vector pLEIN, which expresses EGFP (19) and the neomycin resistance gene on the same bicistronic message (18). Stable, high-expression clones were selected in 800 μg/ml G418 (18). (Bar = 40 μm.)
External Images of Internally Growing B16F0-GFP Tumors.
Metastatic lesions of B16F0-GFP in the brain, bone, liver, and lymph nodes were externally imaged by GFP expression in intact mice after tail vein or portal vein cell injection of B16F0-GFP cells (Figs. 2, 3, and 4).
Figure 2.

External images of murine melanoma (B16F0-GFP) metastasis in brain. Murine melanoma metastases in the mouse brain were imaged by GFP expression under fluorescence microscopy. Clear images of metastatic lesions in the brain can be visualized through the scalp and skull. See Materials and Methods for imaging equipment and procedures. (A) External GFP image of brain metastasis through the scalp and scull of an intact mouse 3 weeks after injection of 106 B16F0-GFP cells in the tail vein. (Bar = 1,280 μm.) (B) GFP image of same area as in A, with skull opened. (Bar = 1,280 μm.) (C) External image obtained of the tumor in the brain of the nude mouse on day 14 after GFP tumor cell injection. (Bar = 1,280 μm.) (D) Same as C, day 19 after injection. (Bar = 1,280 μm.) (E) Same as C and D, day 25 after injection. (Bar = 1,280 μm.) (F) Brain tumor growth curve determined by external images (C–E).
Figure 3.

External images of B16F0-GFP bone metastasis. In the proximal tibia of the left hind leg of C57BL/6 mouse (hair removed). No metastasis can be detected under bright-field microscopy (A). Clear, external images of metastatic lesions of B16F0-GFP in the proximal tibia of the intact mouse were obtained under fluorescence microscopy (B). Time course metastatic growth of B16F0-GFP in the proximal tibia of the intact nude mouse was imaged externally under fluorescence microscopy (C–E). (A) Bright-field microscopy of knee joint of hind leg. (Bar = 640 μm.) (B) Same as A; external fluorescent image of knee joint visualizing extensive melanoma metastasis, day 21 after injection. (Bar = 640 μm.) (C) External image obtained in tibia of nude mouse day 14 after tail vein injection. (Bar = 640 μm.) (D) Same as C, day 20. (Bar = 640 μm.) (E) Same as C and D, day 25. (Bar = 640 μm.) (F) Growth curve of tibia metastatic lesion determined by external images (Fig. 2 C–E).
Figure 4.
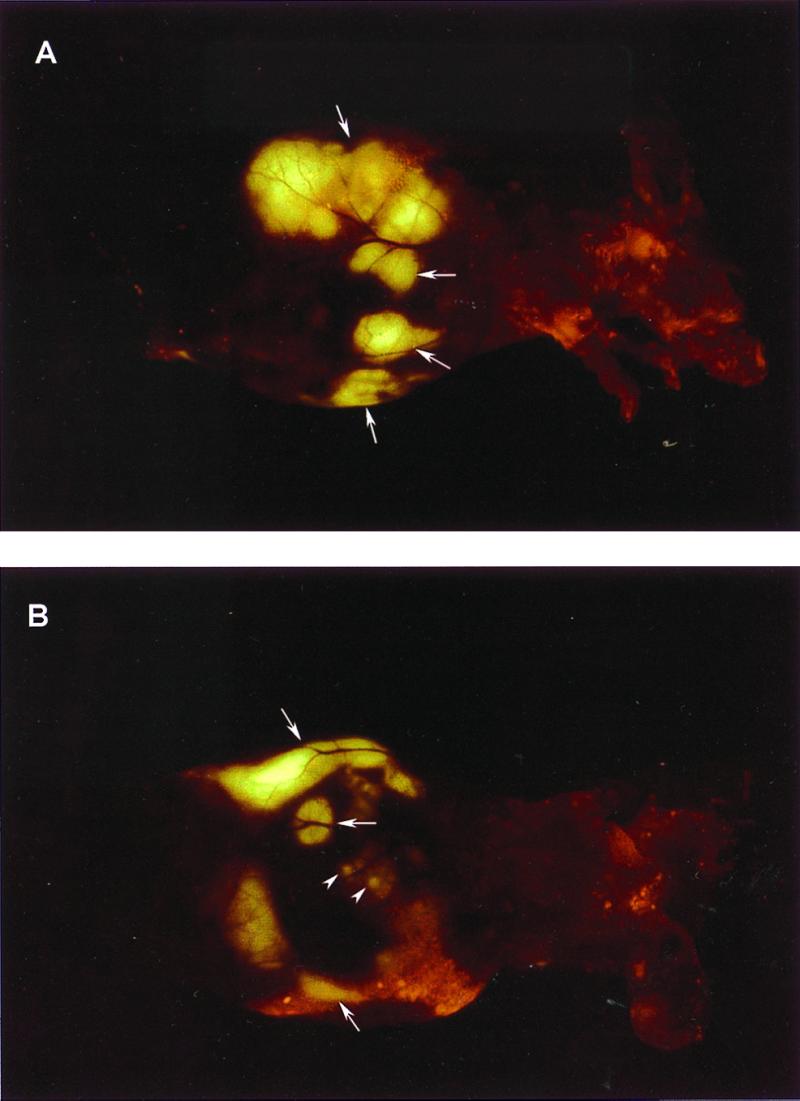
External images of B16F0-GFP colonizing the liver. A metastatic lesion of B16F0-GFP in the liver growing at a depth of 0.8 mm after portal vein injection was externally imaged through the abdominal wall of the intact nude mouse. (A) An external image of multilobe liver metastases of the B16F0-GFP cells (large arrows). (B) An external image of small liver metastatic lesions of approximately 1.5 mm in diameter (small arrows) and other larger metastatic lesions (large arrows).
External Images of Brain Metastasis of B16F0-GFP.
A comparison was made between an external and direct image of a brain metastasis of B16F0-GFP. A fluorescent image (5.5 mm in diameter and 0.8 mm in depth) of B16F0-GFP cells was obtained externally through the scalp and skull of a C57BL/6 mouse (Fig. 2A). The externally acquired image closely matched the image acquired from the open brain after the scalp and skull were removed (Fig. 2B). A series of external fluorescence images of the B16F0-GFP brain tumor in a single animal was obtained from day 14 to day 25 after tail vein injection of B16F0-GFP in a nude mouse. As determined by external imaging, the size of the metastatic lesion grew progressively with time (Fig. 2 C–E). The size of the external image was ≈1.2 mm in diameter at day 14, ≈2.25 mm at day 20, and ≈3.5 mm at day 25.
External Images of Bone Metastasis of B16F0-GFP.
An example of an external fluorescence image of a B16F0-GFP bone metastasis is shown in Fig. 3. The tumor was ≈0.8 mm deep and 2.33 mm in equivalent diameter. The image was acquired at day 21 after tail vein injection. External fluorescent images were acquired throughout the axial skeleton, including the skull, scapula, femur, tibia, and pelvis. Fig. 3 C–E shows a series of external fluorescence images of a tumor in the tibia that were obtained from day 14 to day 25 after tail vein injection of B16F0-GFP in nude mice. The size of the metastatic lesion was ≈0.96 mm in diameter at day 14, ≈1.76 mm at day 20, and ≈3.14 mm at day 25 (Fig. 3F).
External Images of Liver Metastasis of B16FP-GFP.
Metastatic lesions of B16F0-GFP in the nude mouse liver were formed after portal vein injection. A clear external image of multiple metastatic lesions in the liver could be seen through the abdominal wall of the intact mouse at a depth of 0.8 mm (Fig. 4). The image was comparable to the image acquired from the exposed liver (data not shown).
External Images of the AC3488 GFP Human Colon Tumor.
Fluorescent images of the primary colon tumor, multilobe liver metastases, and a skull metastases are shown in Fig. 5. A lateral view of the mouse demonstrates fluorescent liver metastases on the left and right lobes of the liver (Fig. 5A). A cross-section of the mouse, simulating a tomograph, internally localizes the external lateral images of metastatic tumor in the left and right lobes of the liver (Fig. 5B). A dorsal view of the mouse shows the images of metastatic tumors on the caudal portion of these two lobes of the liver (Fig. 5D). The primary colon tumor is imaged in a ventral view of the mouse and suggests local–regional spread (Fig. 5C).
External Images of Skeletal Metastasis of AC3488 GFP.
External fluorescent images of colon tumor metastases throughout the nude mouse skeleton were acquired and compared with direct images of the exposed metastatic lesions (Fig. 6). Fig. 6A shows external, whole-body images of tumors in the skeletal system including the skull, scapula, and spine in a dorsal view of a live, intact nude mouse. External and direct images of the bone metastases were compared. Fig. 6 B–I shows a series of external fluorescent images of metastatic lesions in the skull, ribs, spine, and tibia, which are compared with corresponding images of these skeletal sites acquired after direct exposure of the metastases. It can be seen that the external images correspond highly to the images of the exposed metastatic lesions.
Figure 6.
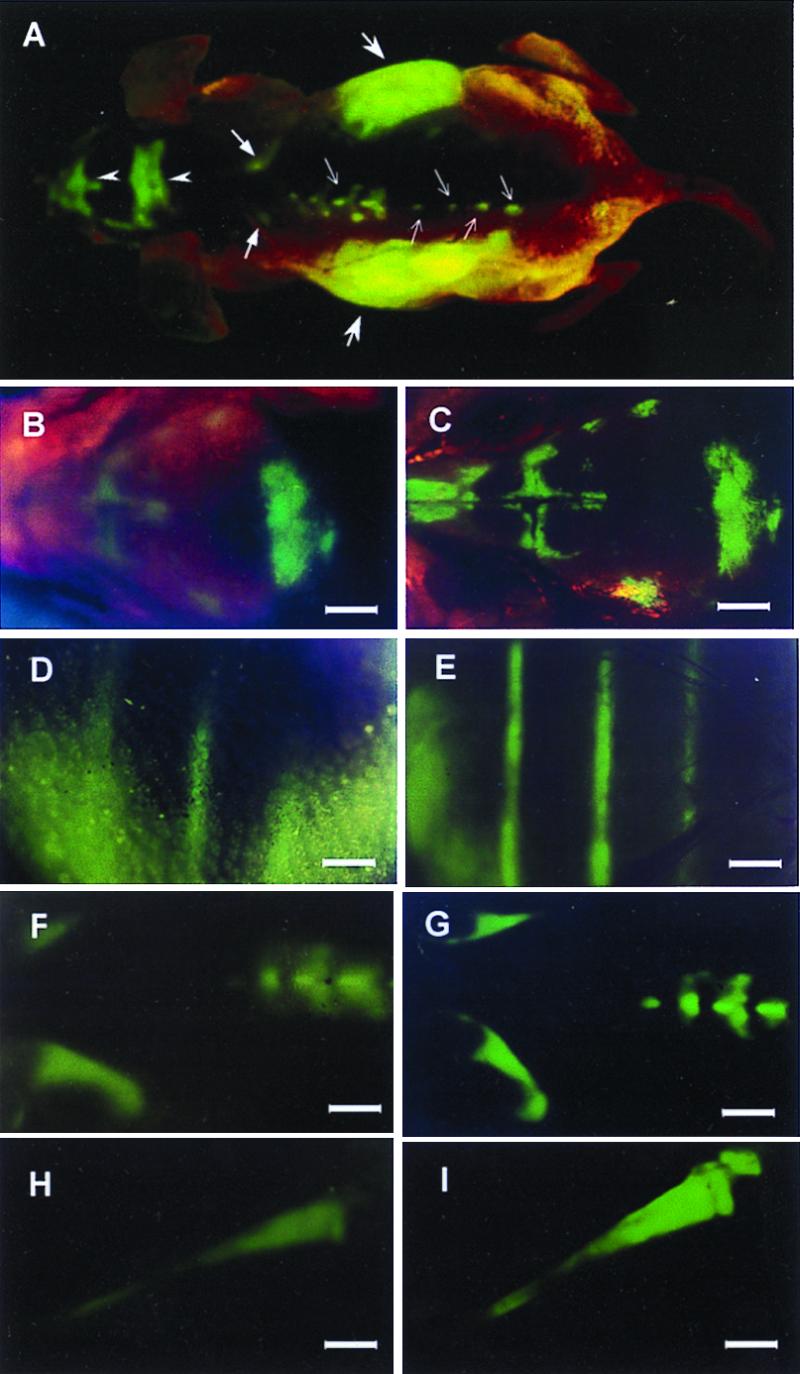
External and internal images of bone metastasis of AC3488 GFP. External fluorescent whole-body images compared with direct images of skeletal metastases. (A) External images of tumors in the skeletal system including the skull (arrow heads), scapula (thick arrows), spine (fine arrows), and liver metastasis (largest arrows) in a dorsal view of live, intact nude mouse. (B–I) Series of external fluorescence images of metastatic lesions in the skull, ribs, spine, and tibia, (B, D, F, and H) compared with corresponding images of the exposed skeletal metastases (C, E, G, and I) (Bars = 1280 μm).
Imaging Sensitivity and Resolution.
GFP-expressing primary and metastatic lesions were considered to be externally measurable if the average fluorescence of the GFP-expressing tumor was at least 20% above the average fluorescence of the surrounding skin. The level of background dorsal and abdominal skin fluorescence of nude mice was in a range of 6–9% of the exposed tumor fluorescence. The intensity of GFP fluorescence of a tumor (1 mm in diameter) growing at a depth of ≈0.8 mm was approximately ≈25% of that of the exposed tumor. The minimum tumor size that could be imaged was a function of depth. The range of minimal size of GFP-expressing tumors that have been imaged externally thus far was from ≈59 μm in diameter at a depth of 0.5 mm to ≈1.86 mm in diameter at a depth of 2.2 mm in various tissues (Table 1).
Table 1.
Minimum-sized fluorescent optical tumor images at increasing depth
| Depth, mm | Minimum-sized tumor imaged, μm (equivalent diameter) |
|---|---|
| 0.5 | 59 |
| 0.6 | 100 |
| 0.7 | 210 |
| 0.8 | 250 |
| 1 | 283 |
| 1.3 | 488 |
| 2.2 | 1,861 |
Tumor images were acquired with a Leica MZ12 microscope coupled with a Hamamatsu C5810 three-chip cooled color charge-coupled device camera. See Materials and Methods for details.
External imaging can provide invaluable real-time data for tracking tumor growth and metastasis formation in live, intact animals. Melanoma metastases in the brain and tibia were imaged over 11 days when they grew from 1.2 to 3.5 mm (Fig. 2 C–F) and from 0.95 to 3.14 mm (Fig. 3 C–F) in equivalent diameter, respectively.
Discussion
The GFP-based fluorescent optical tumor imaging system presents many powerful features. Only the tumors and metastases contain the heritable GFP gene and therefore are selectively imaged with very high intrinsic contrast to other tissues. GFP expression in the tumor cells is stable over indefinite time periods, allowing the quantitative imaging of tumor growth and metastasis formation as well as their inhibition by agents of all types. The very bright GFP fluorescence enables internal tumors and metastases to be observed externally in critical organs such as colon, liver, bone, brain, pancreas (data not shown), and, presumably, breast, lymph nodes, prostate, etc. No contrast agents or other compounds or treatment need to be administered to the animals; only blue light illumination is necessary.
Current sensitivity is limited, in part, by the nonoptimal spectrum of the green GFP fluorescence (520 nm). At this relatively short wavelength, the emitted radiation is strongly scattered by surrounding tissue. However, powerful new techniques of using ultrafast lasers (22), dual photon imaging (23), and ballistic photon imaging (24, 25) may offer large gains in sensitivity, increased depth of detection, and spatial resolution.
The experiments with the B16 melanoma utilized tumor cells that were labeled by external GFP transduction and implanted. However, external labeling is not a necessary limitation on the technique. Recent findings in our laboratory suggest that in situ labeling, with the GFP gene, of tumors growing in vivo is feasible, as shown by the labeling of AC3488 colon tumor in this study (S.H., M.Y., and R.M.H., unpublished data). A wide variety of tumors now can be followed for subsequent tumor growth, spread, and metastases, all reported by inherited GFP expression.
GFP does not appear to be antigenic in either the nude mouse or normal C57B1/6 mouse, because long-term tumor growth and metastasis readily occur in these animals. This new technology will be of very broad use for the understanding of tumor growth and metastasis as well as therapy.
Abbreviation
- GFP
green fluorescent protein
References
- 1.Tearney G J, Brezinski M E, Bouma B E, Boppart S A, Pitris C, Southern J F, Fujimoto J G. Science. 1997;276:2037–2039. doi: 10.1126/science.276.5321.2037. [DOI] [PubMed] [Google Scholar]
- 2.Taubes G. Science. 1997;276:1991–1993. doi: 10.1126/science.276.5321.1991. [DOI] [PubMed] [Google Scholar]
- 3.Baum P R, Brummendorf T H. Q J Nucleic Med. 1998;42:33–42. [Google Scholar]
- 4.Teates C D, Parekh J S. Curr Probl Diagn Radiol. 1993;22:229–226. doi: 10.1016/0363-0188(93)90013-j. [DOI] [PubMed] [Google Scholar]
- 5.Dessureault S. Breast Cancer Res Treat. 1997;45:29–37. doi: 10.1023/a:1005878113826. [DOI] [PubMed] [Google Scholar]
- 6.Pasqualini R, Koivunen E, Ruoslahti R. Nat Biotechnol. 1997;15:542–546. doi: 10.1038/nbt0697-542. [DOI] [PubMed] [Google Scholar]
- 7.Neri D, Carnelmolla B, Nissim A, Leprini A, Querze G, Balza E, Pini A, Tarli L, Halin C, Neri P, et al. Nat Biotechnol. 1997;15:1271–1275. doi: 10.1038/nbt1197-1271. [DOI] [PubMed] [Google Scholar]
- 8.Chambers A F, MacDonald I C, Schmidt E E, Koop S, Morris V L, Khokha R, Groom A C. Cancer Metastasis Rev. 1995;14:279–301. doi: 10.1007/BF00690599. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney T J, Mailander V, Tucker A A, Olomu A B, Zhang W, Cao Y-A, Negrin R S, Contag C H. Proc Natl Acad Sci USA. 1999;96:12044–12049. doi: 10.1073/pnas.96.21.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weissleder R, Tung C H, Mahmood U, Bogdanov A., Jr Nat Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 11.Chishima T, Miyagi Y, Wang X, Yamaoka H, Shimada H, Moossa A R, Hoffman R M. Cancer Res. 1997;57:2042–2047. [PubMed] [Google Scholar]
- 12.Chishima T, Miyagi Y, Wang X, Tan Y, Shimada H, Moossa A R, Hoffman R M. Anticancer Res. 1997;17:2377–2384. [PubMed] [Google Scholar]
- 13.Chishima T, Miyagi Y, Wang X, Baranov E, Tan Y, Shimada H, Moossa A R, Hoffman R M. Clin Exp Metastasis. 1997;15:547–552. doi: 10.1023/a:1018431128179. [DOI] [PubMed] [Google Scholar]
- 14.Chishima T, Miyagi Y, Li L, Tan Y, Baranov E, Yang M, Shimada H, Moossa A R, Hoffman R M. In Vitro Cell Dev Biol Animal. 1997;33:745–747. doi: 10.1007/s11626-997-0151-x. [DOI] [PubMed] [Google Scholar]
- 15.Chishima T, Yang M, Miyagi Y, Li L, Tan Y, Baranov E, Shimada H, Moossa A R, Penman S, Hoffman R M. Proc Natl Acad Sci USA. 1997;94:11573–11576. doi: 10.1073/pnas.94.21.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang M, Hasegawa S, Jiang P, Wang X, Tan Y, Chishima T, Shimada H, Moossa A R, Hoffman R M. Cancer Res. 1998;58:4217–4221. [PubMed] [Google Scholar]
- 17.Yang M, Jiang P, Sun F-X, Hasegawa S, Baranov E, Chishima T, Shimada H, Moossa A R, Hoffman R M. Cancer Res. 1999;59:781–786. [PubMed] [Google Scholar]
- 18.Yang M, Jiang P, An Z, Baranov E, Li L, Hasegawa S, Al-Tuwaijri M, Chishima T, Shimada H, Moossa A R, Hoffman R M. Clin Cancer Res. 1999;5:3549–3559. [PubMed] [Google Scholar]
- 19.Flotte T R, Beck S E, Chesnet K, Potter M, Poirier A, Zolotukhin S. Gene Ther. 1998;5:166–173. doi: 10.1038/sj.gt.3300579. [DOI] [PubMed] [Google Scholar]
- 20.Fu X, Besterman J M, Monosov A, Hoffman R M. Proc Natl Acad Sci USA. 1991;88:9345–9349. doi: 10.1073/pnas.88.20.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun F-X, Sasson A R, Jiang P, An Z, Gamagami R, Li L, Moossa A R, Hoffman R M. Clin Exp Metastasis. 1999;17:41–48. doi: 10.1023/a:1026442321295. [DOI] [PubMed] [Google Scholar]
- 22.Alfano R R, Demos S G, Gayen S K. Ann N Y Acad Sci. 1997;820:248–270. doi: 10.1111/j.1749-6632.1997.tb46200.x. [DOI] [PubMed] [Google Scholar]
- 23.Masters B R, So P T, Gratton E. Ann N Y Acad Sci. 1998;838:58–67. doi: 10.1111/j.1749-6632.1998.tb08187.x. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Perelman L, Dasari R, Feld M. Proc Natl Acad Sci USA. 1997;94:8783–8788. doi: 10.1073/pnas.94.16.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alfano R R, Demos S G, Galland P, Gayen S K, Guo Y, Ho P P, Liang X, Liu F, Wang L, Wang Q Z, et al. Ann N Y Acad Sci. 1998;838:14–28. doi: 10.1111/j.1749-6632.1998.tb08184.x. [DOI] [PubMed] [Google Scholar]