

Abstract
The subtilisin-like prohormone convertases (PCs) contain an essential downstream domain (P domain), which has been predicted to have a β-barrel structure that interacts with and stabilizes the catalytic domain (CAT). To assess possible sites of hydrophobic interaction, a series of mutant PC3–enhanced GFP constructs were prepared in which selected nonpolar residues on the surface of CAT were substituted by the corresponding polar residues in subtilisin Carlsberg. To investigate the folding potential of the isolated P domain, signal peptide–P domain–enhanced GFP constructs with mutated and/or truncated P domains were also made. All mutants were expressed in βTC3 cells, and their subcellular localization and secretion were determined. The mutants fell into three main groups: (i) Golgi/secreted, (ii) ER/nonsecreted, and (iii) apoptosis inducing. The destabilizing CAT mutations indicate that the side chains of V292, T328, L351, Q408, H409, V412, and F441 and nonpolar fragments of the side chains of R405 and W413 form a hydrophobic patch on CAT that interacts with the P domain. We also have found that the P domain can fold independently, as indicated by its secretion. Interestingly, T594, which is near the P domain C terminus, was not essential for P domain secretion but is crucial for the stability of intact PC3. T594V produced a stable enzyme, but T594D did not, which suggests that T594 participates in important hydrophobic interactions within PC3. These findings support our conclusion that the catalytic and P domains contribute to the folding and thermodynamic stability of the convertases through reciprocal hydrophobic interactions.
The proteolytic processing of prohormones and other protein precursors is carried out by a family of calcium-dependent, subtilisin-like serine endoproteases, the prohormone convertases (PCs), that are structurally related to the yeast prohormone processing enzyme kexin (Kex2) (1–3). To date, seven mammalian members of this family have been identified and characterized, including the endoproteinases furin (subtilisin-like proprotein convertase 1; SPC1) (4), PACE4 (SPC4) (5), and the PCs PC2 (SPC2) (6, 7), PC1/PC3 (SPC3) (8, 9), PC4 (SPC5) (10), PC5/PC6 (SPC6) (11), and PC7/PC8/LPC (SPC7) (12–14). Their cleavage sites are usually dibasic pairs, such as KR↓ or RR↓ (15–18). However, basic residues at the P4 and/or P6 position may contribute to substrate recognition or be required for activity, as in the case of furin (15, 19, 20). Thus, minimal consensus sites for furin cleavage are RXK/RR↓ and RXXR↓ (3, 21). All SPCs are synthesized as inactive proenzymes, possessing an N-terminal signal peptide, prodomain, catalytic domain, P domain, and enzyme-specific C-terminal segment (1–3). The upstream prodomain acts as an internal chaperone for the folding of the catalytic domain in the endoplasmic reticulum (ER) (22–25) and also as an intramolecular inhibitor. The activation of the SPCs requires autocatalytic cleavage and removal of the prodomain, a process that usually begins before they exit from the ER (17, 23, 26–30). Recently, the solution structure of the prodomain of PC3 was determined by heteronuclear NMR spectroscopy by Tangrea and colleagues (31). The catalytic domain forms the core structure and contains the well-conserved catalytic triad Asp-His-Ser (32). This domain of the SPCs has shown the greatest similarity (45%) in comparison of sequence alignments of all of the mammalian convertases and kexin (33). Molecular modeling studies have indicated that the catalytic domains of furin and PC3 are closely related to the bacterial subtilisins (19, 34).
A unique feature of the eukaryotic proprotein convertases is the presence of a downstream, well conserved P domain of ≈150 residues. No such homologous sequences have been found in any other subtilase family members or other proteins. The possible functional role of the P domain has been explored by a number of investigators who have shown that the catalytic domain is unstable without it (35–37). Zhou and colleagues (33) demonstrated the importance of the P domain in regulating the stability, calcium ion dependence, and pH dependence of PC3 in domain swapping studies among furin, PC2, and PC3. To better understand the molecular basis of these observations in the absence of a 3D structure, Lipkind et al. (37) have proposed a structural model of the P domain. The modeling study predicts that the P domain is folded as an eight-stranded β-barrel that interacts with the catalytic domain through a hydrophobic patch (37). The variable C-terminal regions of the SPCs are less conserved and mainly play a role in their subcellular routing (1, 22, 38, 39). Both their tissue and subcellular distributions and their substrate specificities influence their normal functions in the body (3).
In the present study, to gain further insight into the protein structure of the SPCs, particularly, the nature of the interaction between the catalytic and P domains, we have carried out systematic mutagenesis studies using enhanced GFP (EGFP)-tagged PC3 (1–616) as a WT control.
Materials and Methods
Construction of Vectors.
Fig. 1 represents the constructs used in this study. cDNA fragments encoding rat PC3 (1–616) was used as a template (33, 38) and subcloned into the pEGFP-N3 expression vector (CLONTECH). The construction of mutations is described in the Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org.
Figure 1.

Constructs of EGFP fusion and mutant proteins used in this study. Full-length or WT PC3 consists of a signal peptide (Pre) and four domains, pro-region (Pro), catalytic domain (Cat), P domain (P), and C-terminal domain. PC3–Asp-616 (PC3D616) is a C-terminally truncated active form of full-length PC3 (amino acids 1–736 of PC3) (38); PC3D616–EGFP is PC3D616-tagged with EGFP (black) via linkage at the N terminus of EGFP and was used as a fully active control in this study. PC3ΔP–EGFP is a P domain deleted (PC3 amino acids 1–453) EGFP-tagged control. Gray-shaded regions represent domains containing point mutations. PC3G593–EGFP and PC3T594–EGFP contain P domains, truncated after G593 or T594, respectively. Sig–PdD616–EGFP consists of a signal peptide (amino acids 1–27), followed by the P domain (amino acids 454–616) tagged with EGFP. In Sig–PdD616–EGFP, the signal peptide and P domain are linked by two added amino acids, glycine (G) and threonine (T). The signal cleavage site was predicted to lie between A27 and G28 in the VKAGTDPR sequence by the SIGNAL P V2.0 server (41, 42). Sig–PdG593–EGFP and Sig–PdT594–EGFP consist of signal peptide, truncated P domain at G593 or T594, respectively, and EGFP.
Cell Culture and Transfection.
βTC3 cells were routinely maintained in DMEM supplemented with 10% FCS (Life Technologies, Grand Island, NY). Twenty-four hours after seeding on a round coverslip in the bottom of a six-well plate, cells were transiently transfected with 500 ng of EGFP expression vectors by using Effectane Transfection Regent according to the manual (Qiagen, Chatsworth, CA).
Immunostaining for Endogenous Insulin.
Fixed cells were washed three times with PBS and blocked by 5% normal donkey serum (Jackson ImmunoResearch) at 4°C overnight. Cells were then incubated with a 1:1,000 dilution of guinea pig anti-insulin antibody (DAKO) at 4°C overnight and incubated with a 1:500 dilution of Cy3-conjugated donkey anti-guinea pig IgG (Jackson ImmunoResearch) for 30 min at room temperature before fluorescence microscopy.
Detection of Apoptosis.
To observe fragmentated nuclei of apoptotic cells, the nuclei of fixed cells were stained with Hoechst 33342. For detection of 3′ OH DNA ends, a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay, DeadEnd Colorimetric TUNEL System (Promega) was performed according to the manufacturer's instructions. For a positive control, βTC3 cells were incubated in the culture medium with 10 μg/ml tunicamycin (Sigma-Aldrich).
Western Blotting.
Forty to 48 hours after transfection, medium was replaced with complete serum-free medium and incubated for 20 h. The cell extracts and 40× concentrated medium were loaded on 7.5% SDS/PAGE and then transferred onto nitrocellulose membranes by electroblotting. Immunoblotting was conducted with a 1:400 dilution of rabbit anti-GFP antibody (CLONTECH).
Modeling Study.
insight ii (Biosym Technologies, San Diego) was used for modeling studies. The x-ray structures of subtilisin Carlsberg and γ crystallin were used as structural templates for the catalytic and P domains, respectively (37).
Results
The P Domain Is Required for PC3 Folding and Secretion via the Secretory Pathway.
To verify the importance of the P domain for the folding of proPC3 in the context of EGFP, expression of PC3ΔP–EGFP was compared with PC3D616–EGFP (truncated WT control) in terms of both subcellular localization and secretion in βTC3 cells. The fates of these proteins differed significantly. PC3D616–EGFP was seen to colocalize in the Golgi and secretory granules near the cell surface (Fig. 2Aa), along with endogenous proinsulin (Fig. 2Ae). Moreover, PC3D616–EGFP was secreted into the medium (Fig. 1B). In contrast to PC3D616–EGFP, PC3ΔP–EGFP remained in the ER (Fig. 2Ab), did not colocalize with endogenous insulin (Fig. 2Af), and was not detected in the medium (Fig. 2B). These data reveal that PC3D616–EGFP can be sorted normally into the regulated secretory pathway, whereas PC3ΔP–EGFP cannot, but instead accumulates in the ER. This finding is thus consistent with earlier observations that PC3 cannot fold without a P domain (33, 36, 40).
Figure 2.

The P domain is required for PC3 progression within the secretory pathway. (A) Analysis of subcellular localization by fluorescence microscopy. PC3D616–EGFP (a) and PC3ΔP–EGFP (b) expressed in βTC3 cells were observed as green. Endogenous insulin was identified by anti-insulin antibody (Cy3) (c and d) (red), and colocalization of EGFP-tagged proteins and endogenous insulin was observed as yellow in merged images (e and f). Golgi–enhanced cyan fluorescent protein and ER-enhanced yellow fluorescent protein were used as markers for Golgi (g) and ER (h), respectively. PC3D616–EGFP was predominantly localized in Golgi and secretory granules near the cell surface along with insulin, whereas PC3ΔP–EGFP was localized in the ER. (Magnification: ×800.) (B) Analysis of the expression and secretion of EGFP-tagged proteins by Western blotting using anti-GFP antibody. PC3D616–EGFP was detected as a 94-kDa band in both cell extract and medium. PC3ΔP–EGFP was detected in cell extract as a 75-kDa band, but was not detected in the medium.
Effects of Mutagenesis of Hydrophobic Residues on the Surface of the Catalytic Domain.
Comparative analysis of the surface residues of the structurally conserved framework of subtilisin Carlsberg and PC3 revealed a region where charged amino acid residues in subtilisin are replaced by hydrophobic residues in PC3 and related convertases (37). The acidic residues, Asp-172, Glu-195, and Glu-197, of subtilisin are substituted by Val, Ser, and Leu residues, respectively in the case of PC3, whereas the basic residues Lys-170, Arg-247, Arg-249, and Lys-265 are changed to Ile, Leu, Val, and Phe, respectively. These residues are clustered on the surface so as to form a hydrophobic patch unique to the surfaces of the SPCs. In PC3, it is formed by residues V293, T328, I326, S349, L351, V412, F441, W413, Q408, and L410 and very likely interacts with its P domain.
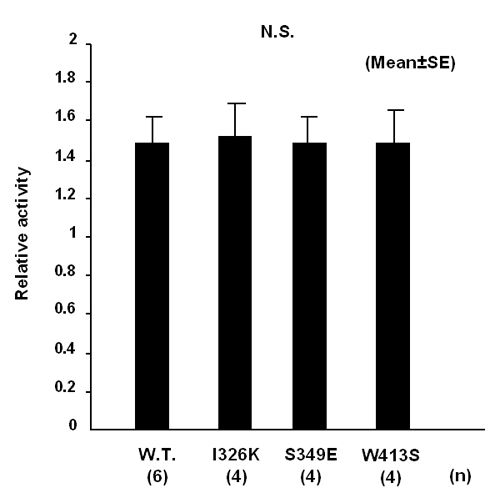
To clarify the prediction that this hydrophobic patch is not located on the external surface of PC3, but instead participates in important intramolecular interactions within the core of the PC3 holoenzyme, we have mutated these hydrophobic residues to the corresponding charged or polar residues found in subtilisin Carlsberg and expressed these mutants in βTC3 cells. The subcellular localizations of mutants I326K, S349E, and W413S were similar to that of WT (PC3D616–EGFP). These all progressed normally to the Golgi region, indicating that their folding was not disrupted; thus the side chains of these residues are probably directed outside the hydrophobic patch. However, this was not the case for the other seven mutants, which all were retained in the ER similarly to PC3ΔP–EGFP (Fig. 3A). As expected, Western blots showed that the mutants that exhibited the Golgi pattern were all secreted into the medium, whereas none of the ER pattern mutants were secreted (Fig. 3B). Moreover, in the case of the nonsecreted mutants, L410R, V412S, and F441K, the number of EGFP-expressing cells in the transfected cultures was much lower than in the case of the other mutants, and almost all EGFP-positive cells expressing these mutants were rounded and small, and their nuclear area could not be clearly delineated by fluorescence microscopy. Fragmentation of nuclei was detected by nuclear staining (Fig. 3C), and apoptotic cells were visualized in increased numbers by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling assay (Fig. 3D) in these cultures. Therefore, based on their subcellular localizations and secretion, these catalytic domain mutants could be classified into three groups (Table 1). Three mutants (I326K, S349E, and W413S) were normally transported and secreted (Golgi/secreted group), four mutants (V292Y, T328D, L351E, and Q408Y) were retained in the ER (ER/nonsecreted group), and three mutants (L410R, V412S, and F441K) tended to cause cell death (apoptosis-inducing group). Additionally, secreted mutant proteins, I326K, S349E, and W413S had levels of enzyme activity similar to that of WT controls (see Fig. 8, which is published as supporting information on the PNAS web site).
Figure 3.

Mutation of residues comprising the predicted hydrophobic patch on the surface of the catalytic domain. Ten positions mutated to correspond to their homologous residues in subtilisin Carlsberg (37) were studied. (A) Analysis of subcellular localization by fluorescence microscopy. (B) Western blotting using anti-GFP antibody. From the results of studies of subcellular localization and secretion, those mutants were classified into three groups: Golgi/secreted (I326K, S349E, and W413S), ER/nonsecreted (V292Y, T328D, L351E, and Q408Y), and cell death (apoptosis-inducing) (L410R, V412S, and F441K). In the cell death group, fragmentation of nuclei (arrows) was detected by nuclear staining using Hoe33342 (C) and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling-positive cells were stained (dark brown) (D). In the latter experiment, a positive control for apoptosis was provided by treatment of WT PC3 with tunicamycin (Upper Middle). (Magnifications: A, ×800; C, ×400; D, ×200.)
Table 1.
Classification of the fates of EGFP-tagged proteins expressed in this study based on their subcellular localization and secretion
| Result | Golgi/secreted | ER/nonsecreted | Apoptosis-inducing |
|---|---|---|---|
| I | PC3D616–EGFP | PC3ΔP-EGFP | |
| II | I326K, S349E, W413S | V292Y, T328D, L351E, Q408Y, H409D, H409K | L410R, V412S, F441K |
| III | K588A, K588D, L589A | L589D, L591D, H592A | |
| I590A, I590D, I590K | G593A, G593D, G593R | ||
| I590R, L591A, H592D | T594A, T594D | ||
| T594V | |||
| IV | PC3T594–EGFP | PC3G593-EGFP | |
| Sig-PdD616–EGFP | |||
| Sig-PdT594–EGFP | |||
| Sig-PdG593–EGFP |
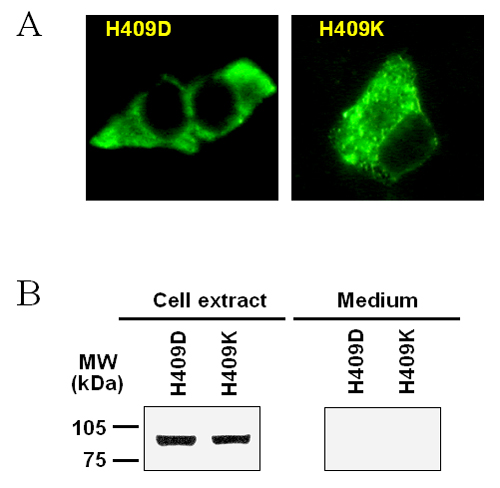
These results are thus consistent with the notion that the mutated residues in the ER/nonsecreted and apoptosis-inducing groups are important for the correct folding of the protein. Therefore, Val-292, Thr-328, Leu-351, Gln-408, Leu-410, Val-412, and Phe-441, which do not tolerate substitutions by polar amino acid residues, as are present in subtilisin, must play a special role in the structural organization of PC3. The most reasonable conclusion is that they form a compact and flat hydrophobic patch on the surface of the catalytic domain, which engages the P domain during folding of the proenzyme. In addition, more careful analysis of surrounding amino acid residues indicated that the side chain of His-409 might participate in this patch, along with nonpolar portions of the side chains of both Arg-405 and Trp-413. Modeling also indicated that the side chain of Leu-410 is not likely to be part of this surface, but is directed toward the interior. Two additional mutations (H409D and H409K) were also both ER-retained (see Fig. 9, which is published as supporting information on the PNAS web site). This finding suggested that His-409 also projects toward the P domain interface. The revised topography of this hydrophobic patch on the surface of PC3, based on these experimental data, is shown in Fig. 4.
Figure 4.

The topography of the hydrophobic surface of the catalytic domain. The space-filling residues shown comprise a hydrophobic patch on the surface of the catalytic domain of PC3, which is postulated to interact with the P domain. An earlier model of this hydrophobic surface (37) was revised based on the results of the present mutagenesis study. The aliphatic portions of the side chains of R405 and W413 also may contribute to the hydrophobic patch, whereas I326 and S349 are excluded. Residue L410 was also excluded from this image because our modeling results indicated that the L410 side chain is directed toward the interior of the catalytic domain. The numbers in parentheses represent positions in subtilisin Carlsberg.
Mutagenesis of Neighboring Residues Near the C Terminus of the P Domain.
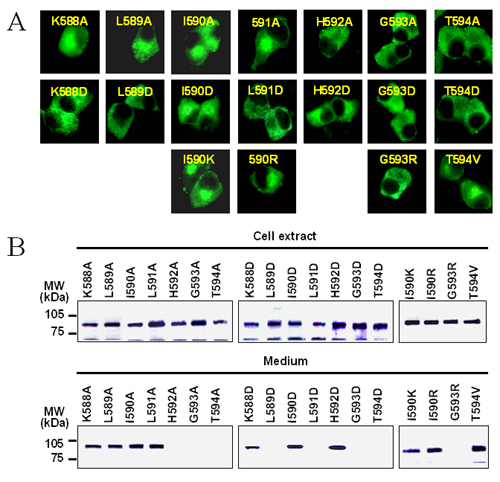
Previous studies by Zhou and colleagues (33) have revealed the importance of several conserved amino acids in the C-terminal region of the P domain (His-592, Gly-593, and Thr-594). These play a crucial role not only in sustaining the autocatalytic activity but also in the folding and secretion of PC3 (33). Jackson et al. (45) have also reported a patient with a phenotype of defective prohormone processing caused by compound heterozygosity for defects in the PC3 gene, including a Gly-483–Arg missense allele and an allele with an A to C transversion at position +4 of the donor splice site of intron 5. In their report, PC3 Gly-483–Arg (which corresponds to Gly-593 in rat PC3) was shown be retained in the ER. Therefore, to more clearly understand how this region of the P domain stabilizes the structure of PC3, a systematic mutagenesis study in this region was performed. We individually mutated all of the residues from Lys-588 to Thr-594 to alanine (A) or aspartic acid (D). The results of this analysis (see Fig. 10, which is published as supporting information on the PNAS web site) revealed that Lys-588, Ile-590, and His-592 could be substituted with the negatively charged residue, aspartic acid, whereas Leu-589 and Leu-591 could not. This region previously was predicted to form a β-strand at the end of the proposed β-barrel structure of the P domain (37). The alternating acceptance of substitutions by charged residues within this segment indeed coincides with the alternating directions of the side chains in a β-strand. Moreover, Lys-588, Ile-590, and Leu-591 could be substituted with alanine, but His-592, Gly-593, and Thr-594 could not (Fig. 5). These data indicate that the side chains of Leu-589 and Leu-591 project toward the interior of the P domain, whereas Lys-588, Ile-590, and His-592 project outwardly, thus supporting the previous predictions (37). The three external residues in this β-strand probably do not interact with the hydrophobic patch on the surface of the catalytic domain, because they are easily substituted by the negatively charged aspartic acid. The next residue in the sequence, Gly-593, cannot be substituted by any residues with a Cβ atom, which strongly indicates that Gly-593 forms a β-turn at the end of the last β-strand of the P domain. However, the essential Thr-594 residue could be substituted with the bulky hydrophobic residue, valine, or with serine, but not with aspartic acid or asparagine. These findings suggest that Thr-594 may play an important role in specific hydrophobic interactions with the catalytic domain that stabilize the PC3 structure.
Figure 5.

Mutations near the carboxyl terminus of the P domain. ER-retained and tolerated mutations are shown by red and blue, respectively. Highlighted sequence (yellow) is in the region of the last β-strand of the P domain predicted by the Chou and Fasman method (43, 44), and arrows indicate hydrophobic residues that are directed toward the P domain's interior, according to the previous modeling study (37). The mutations marked by * have already been reported (33, 45), and underlined mutations were reconfirmed in this study. The spatial structure of this β-strand is given below, where the yellow vectors represent alternative directions of the side chains. W587, L589, and L591 are inside the protein and K588, I590, and H592 are outside. G593 forms a β-turn next to the β-strand.
Effect of C-Terminal Truncations of the P Domain.
To investigate whether T594 is directed toward the hydrophobic core of PC3 or the hydrophobic core of the P domain itself, further truncation studies of the P domain were performed (Fig. 6). Initially, we confirmed that the P domain can fold in the absence of the catalytic domain, using the Sig–PdD616–EGFP construct. Then, the C-terminal region of the P domain was truncated up to T594 not only in the WT control, PC3D616–EGFP, but also in Sig–PdD616–EGFP. As expected, Sig–PdD616–EGFP was sorted into secretory pathway, which indicates that the P domain (amino acids 454–616) can properly fold without the catalytic domain. Truncation after Thr-594 had no effect on folding or secretion in both control and Sig–PdD616–EGFP. But, interestingly, truncation after Gly-593 was disruptive in the WT control, but not in Sig–PdD616–EGFP, lacking the catalytic domain. These results indicate that Thr-594 is necessary for the folding of the PC3 holoenzyme precursor, but not for the folding of the isolated P domain. From this finding, we believe that Thr-594 is normally oriented toward the interface with the catalytic domain in PC3. Taken together with the results of the former mutagenesis study, Thr-594 evidently plays an important stabilizing role and also serves as the functional C terminus of the P domain.
Figure 6.

Truncation study of P domain. Subcellular localization (A) and expression and secretion assessed by Western blotting (B) of PC3T594–EGFP (a), PC3G593–EGFP (b), Sig–PdD616–EGFP (c), Sig–PdT594–EGFP (d) and Sig–PdG593–EGFP (e) in βTC3 cells are shown. PC3T594–EGFP and PC3G593–EGFP are C-terminally truncated forms of PC3D616–EGFP (active control). Among these truncations, only lack of T594 in PC3 (b) resulted in ER retention. Sig–PdD616–EGFP could be sorted into Golgi and secreted into the medium, where it was detected as a 45-kDa band; however, in the absence of the catalytic domain, truncation after G593 of the P domain (e) did not prevent secretion. (Magnification: A, ×800.)
Discussion
Recently, GFP has been widely used in the field of cell biology to monitor many cellular processes, including the secretory pathway (46, 47). Therefore, in this study, we have used EGFP protein as a downstream reporter domain to follow the progress of PC3 derivatives and mutants through the secretory pathway. The purpose of this study was to assess the role of mutual hydrophobic interactions between the P and catalytic domains for the folding of PC3. The most notable difference in primary structures between the catalytic domains of the SPCs and subtilisins is the large increase in number of negatively charged residues, glutamic acid and aspartic acid, clustered on the surface of the SPC catalytic domains in the substrate binding region (37). We propose that this charge imbalance may lead to instability in the catalytic domain, which is offset by interaction of the P domain with the catalytic domain to provide stabilization. Indeed, several truncation experiments have shown the P domains are required for the stable folding of the SPCs (33, 36, 40). In those prior studies, P domain-deleted convertases were expressed in cells, but were unable to traverse the secretory pathway, an indication of misfolding. In agreement with those previous experiments, PC3ΔP–EGFP in our experiments was not sorted into the secretory pathway and secreted normally, but was retained in the ER, although PC3D616–EGFP, a truncated but fully functional WT control, was sorted normally into secretory granules and secreted as an active enzyme. These findings demonstrated that PC3ΔP–EGFP also is unable to fold normally and this defect is not altered by the presence of the EGFP tag.
In the previous modeling study of Lipkind et al. (37), a conserved patch of hydrophobic residues was identified on the surface of the catalytic domain on the side away from the active site and it was posited that this patch is a likely candidate for interaction with the independently folded β-barrel structure proposed for the P domain. Ten amino acids were predicted to form a patch, which could interact with the P domain (37). The present mutagenesis study in which these amino acid residues have been reverted to their polar counterparts in subtilisin Carlsberg has resulted in a more precise identification of residues in the patch that influence the folding of PC3. Some of these mutants were retained in ER and others induced apoptosis, whereas a few were well tolerated. Interestingly, the tolerated mutations were arranged around the periphery of the predicted hydrophobic patch, whereas the nontolerated mutations were located within its central region. Several mutations that caused apoptosis may have induced the unfolded protein response more strongly than the other misfolding mutants, suggesting the possible existence of different aberrant folding pathways or states (48).
We also mutated the highly conserved carboxyl-terminal region of the P domain (residues 588–594). We observed an alternating pattern of tolerance of substitutions by aspartic acid residues with respect to protein secretion (secreted/not secreted) in the sequence 588–592, which supports the prediction that this region forms a β-strand in the folded P domain structure (37). At the same time our data show, that if residues Leu-589 and Leu-591 participate in the formation of the hydrophobic core of the P domain, the other residues of this β-strand (Lys-588, Ile-590, and His-592) are likely to be directed toward the exterior.
Moreover, our results confirm that G593R is retained in the ER, a finding consistent with the occurrence of an inactivating mutation in a human subject (45). Zhou et al. (33) presented data showing that the shortest active form of PC3 was PC3T594 and that threonine could not replace His-592. Their results were suggestive of an important function of these terminal residues of the P domain. The additional mutation, T594V, along with T594S (33), is most informative, as both mutants were tolerated. The fact that Thr-594 can be successfully substituted by the bulky hydrophobic residue, valine (but not by aspartic acid), indicates that the threonine side chain very likely participates in an important stabilizing hydrophobic interaction within PC3, because we have shown that Thr-594 itself is not important for the folding of the P domain. Both Gly-593 and Thr-594 are completely conserved among the mammalian SPCs (37). Interestingly, Gly-593 could not be substituted with alanine or aspartic acid. The absence of a side chain in glycine confers great conformation flexibility to the polypeptide backbone. We propose that Gly-593 interrupts the last β-strand at the C terminus of the P domain. These findings indicate that Thr-594 is crucial for the stable folding of the entire proenzyme, rather than just the boundary region at the end of the P domain.
The downstream C-terminal segment beyond Thr-594 appears to play a role in directing PC3 into the regulated secretory pathway. Zhou et al. (38) have shown that PC3 truncated at D616 is sorted predominantly into the constitutive pathway and is enzymatically active. This naturally occurring truncated form, which was fused with EGFP and used as a WT control (PC3D616–EGFP) in our study, also is active, but instead is sorted into the regulated secretory pathway, as indicated by its colocalization with endogenous insulin-containing granules. It thus appears that tagging PC3D616 with EGFP restores normal sorting behavior to the truncated enzyme. However, interestingly, deletion of the segment from Ser-595 to Asp-616 caused a decrease in high-intensity EGFP signals near the surface of the cell membrane; for example, compare PC3D616–EGFP to PC3D594–EGFP, and SigPd–D616–EGFP to SigPd–T594–EGFP and G593–EGFP in Figs. 2A and 6A. This finding suggests that the segment from Ser-595 to Asp-616 in PC3, which is also well conserved among various species, may influence its sorting behavior, in concert with residues after D616.
We conclude from these studies that the P domain contributes to the folding and thermodynamic stability of PC3 through specific hydrophobic interactions with the catalytic domain and that Thr-594 is the precise boundary between the P domain and the C-terminal segment (Fig. 7). Thr-594 is crucially important for the correct folding of the holoenzyme and participates in hydrophobic interactions inside the PC3 structure. On the basis of these experimental data, we have revised the dimensions of the hydrophobic patch on the surface of the catalytic domain that is predicted to interact with the hydrophobic surface of the P domain. We also conclude that amino acid residues, near the C-end of the P domain (587–593) form a β-strand.
Figure 7.

Model of the hydrophobic surface of the P domain that is postulated to interact with the catalytic domain. The hydrophobic residues on the external sides of β-strands 5 and 6 of the β-barrel of the P domain, shown by large space-filling images, could participate in hydrophobic interactions with the catalytic domain. The side chains of β-strand 8 of the P domain, which do not participate in such interactions, and are shown by small balls and sticks.
Supplementary Material
Acknowledgments
We thank Jie Wang and Arunangsu Dey for helpful discussions of this work and Rosie Ricks for expert assistance in preparing this manuscript. This work was supported by the Howard Hughes Medical Institute and U.S. Public Health Service Grants DK13914 and 20595.
Abbreviations
- PC
prohormone convertase
- SPC
subtilisin-like proprotein convertase
- EGFP
enhanced GFP
- ER
endoplasmic reticulum
References
- 1.Steiner D F. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 2.Zhou A, Webb G, Zhu X, Steiner D F. J Biol Chem. 1999;274:20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]
- 3.Thomas G. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van de Ven W J M, Voorberg J, Fontijn R, Pannekoek H, Van den Ouweland A M W, Van Duijnhoven H L P, Roebroek A J M, Siezen R J. Mol Biol Rep. 1990;14:265–275. doi: 10.1007/BF00429896. [DOI] [PubMed] [Google Scholar]
- 5.Kiefer M C, Tucker J E, Joh R, Landsberg K E, Saltman D, Barr P J. DNA Cell Biol. 1991;10:757–769. doi: 10.1089/dna.1991.10.757. [DOI] [PubMed] [Google Scholar]
- 6.Smeekens S P, Steiner D F. J Biol Chem. 1990;265:2997–3000. [PubMed] [Google Scholar]
- 7.Seidah N G, Mattei M G, Gaspar L, Benjannet S, Mbikay M, Chretien M. Genomics. 1991;11:103–107. doi: 10.1016/0888-7543(91)90106-o. [DOI] [PubMed] [Google Scholar]
- 8.Smeekens S P, Avruch A S, LaMendola J, Chan S J, Steiner D F. Proc Natl Acad Sci USA. 1991;88:340–344. doi: 10.1073/pnas.88.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidah N G, Marcinkiewicz M, Benjannet S, Gaspar L, Beaubien G, Mattei M G, Lazure C, Mbikay M, Chretien M. Mol Endocrinol. 1991;5:111–122. doi: 10.1210/mend-5-1-111. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama K, Kim W-S, Torii S, Hosaka M, Nakagawa T, Ikemizu J, Baba T, Murakami K. J Biol Chem. 1992;267:5897–5900. [PubMed] [Google Scholar]
- 11.Nakagawa T, Hosaka M, Torii S, Watanabe T, Murakami K, Nakayama K. J Biochem (Tokyo) 1993;113:132–135. doi: 10.1093/oxfordjournals.jbchem.a124015. [DOI] [PubMed] [Google Scholar]
- 12.Seidah N G, Hamelin J, Mamarbachi M, Dong W, Tardos H, Mbikay M, Chretien M, Day R. Proc Natl Acad Sci USA. 1996;93:3388–3393. doi: 10.1073/pnas.93.8.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruzzaniti A, Goodge K, Jay P, Taviaux S A, Lam M H D, Berta P, Martin T J, Moseley J M, Gillespie M T. Biochem J. 1996;314:727–731. doi: 10.1042/bj3140727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meerabux J, Yaspo M-L, Roebroek A J, Van den Ven M J M, Lister T A, Young B D. Cancer Res. 1996;56:448–454. [PubMed] [Google Scholar]
- 15.Steiner D F, Smeekens S P, Ohagi S, Chan S J. J Biol Chem. 1992;267:23435–23438. [PubMed] [Google Scholar]
- 16.Steiner D F, Bell G I, Tager H S, Rubenstein A H. In: Endocrinology. DeGroot L, editor. Philadelphia: Saunders; 1995. pp. 1296–1328. [Google Scholar]
- 17.Rouille Y, Duguay S, Lund K, Furuta M, Gong Q, Lipkind G, Oliva A, Jr, Chan S J, Steiner D F. Front Neuroendocrinol. 1995;16:322–361. doi: 10.1006/frne.1995.1012. [DOI] [PubMed] [Google Scholar]
- 18.Steiner D F, Rouille Y, Gong Q, Martin S, Carroll R, Chan S J. Diabetes Metab. 1996;22:94–104. [PubMed] [Google Scholar]
- 19.Lipkind G, Gong Q, Steiner D F. J Biol Chem. 1995;270:13277–13284. doi: 10.1074/jbc.270.22.13277. [DOI] [PubMed] [Google Scholar]
- 20.Furuta M, Carroll R, Martin S, Swift H, Ravazzola M, Orci L, Steiner D F. J Biol Chem. 1998;273:3431–3437. doi: 10.1074/jbc.273.6.3431. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama K. Biochem J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creemers J W, Jackson R S, Hutton J C. Semin Cell Dev Biol. 1998;9:3–10. doi: 10.1006/scdb.1997.0195. [DOI] [PubMed] [Google Scholar]
- 23.Seidah N, Mbikay M, Marcinkiewicz M, Chrétien M. In: Proteolytic and Cellular Mechanisms in Prohormone Processing. Hook V, editor. Georgetown, TX: Landes; 1998. pp. 49–76. [Google Scholar]
- 24.Hu Z, Zhu X, Jordan F, Inouye M. Biochemistry. 1994;33:562–569. doi: 10.1021/bi00168a022. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Ohta Y, Jordan F, Inouye M. Nature. 1989;339:483–484. doi: 10.1038/339483a0. [DOI] [PubMed] [Google Scholar]
- 26.Anderson E, VanSlyke J, Thulin C, Jean F, Thomas G. EMBO J. 1997;16:1508–1518. doi: 10.1093/emboj/16.7.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Bie I, Marcinkiewicz M, Malide D, Lazure C, Nakayama K, Bendayan M, Seidah N G. J Cell Biol. 1996;135:1261–1275. doi: 10.1083/jcb.135.5.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van de Loo J-W H, Creemers J W, Bright N A, Young B D, Roebroek A J, Van de Ven W J. J Biol Chem. 1997;272:27116–27123. doi: 10.1074/jbc.272.43.27116. [DOI] [PubMed] [Google Scholar]
- 29.Munzer J S, Basak A, Zhong M, Mamarbachi A, Hamelin J, Savaria D, Lazure C, Hendy G N, Benjannet S, Chretien M, Seidah N G. J Biol Chem. 1997;272:19672–19681. doi: 10.1074/jbc.272.32.19672. [DOI] [PubMed] [Google Scholar]
- 30.Mains R E, Berard C A, Denault J-B, Zhou A, Johnson R C, Leduc R. Biochem J. 1997;321:587–593. doi: 10.1042/bj3210587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tangrea M A, Bryan P N, Sari N, Orban J. J Mol Biol. 2002;320:801–812. doi: 10.1016/s0022-2836(02)00543-0. [DOI] [PubMed] [Google Scholar]
- 32.Siezen R J, Leunissen J A M. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou A, Martin S, Lipkind G, LaMendola J, Steiner D F. J Biol Chem. 1998;273:11107–11114. doi: 10.1074/jbc.273.18.11107. [DOI] [PubMed] [Google Scholar]
- 34.Siezen R J, de Vos W M, Leunissen J A M, Dijkstra B W. Protein Eng. 1991;4:719–737. doi: 10.1093/protein/4.7.719. [DOI] [PubMed] [Google Scholar]
- 35.Creemers J W, Usac E F, Bright N A, Van de Loo J-W, Jansen E, Van de Ven W J M, Hutton J C. J Biol Chem. 1996;271:25284–25291. doi: 10.1074/jbc.271.41.25284. [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Muller L, Mains R E, Lindberg I. J Biol Chem. 1998;273:1158–1164. doi: 10.1074/jbc.273.2.1158. [DOI] [PubMed] [Google Scholar]
- 37.Lipkind G, Zhou A, Steiner D. Proc Natl Acad Sci USA. 1998;95:7310–7315. doi: 10.1073/pnas.95.13.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou A, Paquet L, Mains R E. J Biol Chem. 1995;270:21509–21516. doi: 10.1074/jbc.270.37.21509. [DOI] [PubMed] [Google Scholar]
- 39.Jutras I, Seidah N G, Reudelhuber T L, Brechler V. J Biol Chem. 1997;272:15184–15188. doi: 10.1074/jbc.272.24.15184. [DOI] [PubMed] [Google Scholar]
- 40.Gluschankof P, Fuller R S. EMBO J. 1994;13:2280–2288. doi: 10.1002/j.1460-2075.1994.tb06510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claros M G, Brunak S, von Heijne G. Curr Opin Struct Biol. 1997;7:394–398. doi: 10.1016/s0959-440x(97)80057-7. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 43.Chou P Y, Fasman G D. Biochemistry. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 44.Chou P Y, Fasman G D. Annu Rev Biochem. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 45.Jackson R S, Creemers J W, Ohagi S, Raffin-Sanson M L, Sanders L, Montague C T, Hutton J C, O'Rahilly S. Nat Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 46.El Meskini R, Jin L, Marx R, Bruzzaniti A, Lee J, Emeson R, Mains R. Endocrinology. 2001;142:864–873. doi: 10.1210/endo.142.2.7929. [DOI] [PubMed] [Google Scholar]
- 47.Molinete M, Lilla V, Jair R, Joyce P B, Gorr S U, Ravazzola M, Halban P A. Diabetologia. 2000;43:1157–1164. doi: 10.1007/s001250051507. [DOI] [PubMed] [Google Scholar]
- 48.Fewell S W, Travers K J, Weissman J S, Brodsky J L. Annu Rev Genet. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}