

Abstract
APOBEC3G and APOBEC3F restrict human immunodeficiency virus type 1 (HIV-1) replication in vitro through the induction of G→A hypermutation; however, the relevance of this host antiviral strategy to clinical HIV-1 is currently not known. Here, we describe a population level analysis of HIV-1 hypermutation in near-full-length clade B proviral DNA sequences (n = 127). G→A hypermutation conforming to expected APOBEC3G polynucleotide sequence preferences was inferred in 9.4% (n = 12) of the HIV-1 sequences, with a further 2.4% (n = 3) conforming to APOBEC3F, and was independently associated with reduced pretreatment viremia (reduction of 0.7 log10 copies/ml; P = 0.001). Defective vif was strongly associated with HIV-1 hypermutation, with additional evidence for a contribution of vif amino acid polymorphism at residues important for APOBEC3G-vif interactions. A concurrent analysis of APOBEC3G polymorphism revealed this gene to be highly conserved at the amino acid level, although an intronic allele (6,892 C) was marginally associated with HIV-1 hypermutation. These data indicate that APOBEC3G-induced HIV-1 hypermutation represents a potent host antiviral factor in vivo and that the APOBEC3G-vif interaction may represent a valuable therapeutic target.
APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G) and the closely related APOBEC3F are recently identified anthropoid-specific proteins that restrict human immunodeficiency virus (HIV) type 1 (HIV-1) replication by deaminating cytosine residues in intermediary single-stranded HIV DNA. This host antiviral strategy thereby introduces DNA editing errors into the retroviral genome sequence, resulting in the fixation of an inordinate number of proviral HIV DNA guanine-to-adenine (G→A) substitutions referred to as hypermutation (10, 20, 43). Previous studies have defined polynucleotide motifs within single-stranded DNA that are preferentially targeted by APOBEC3G (resulting in proviral DNA GG→AG substitutions [substituted bases are underlined and italicized]) and APOBEC3F (GA→AA) (2, 16, 36). These studies have also identified HIV-1 viral infectivity protein (vif) as the principal viral factor that counteracts APOBEC3-mediated DNA editing by promoting the degradation of APOBEC3-vif complexes via the proteasomal pathway (6, 21, 42). Recent in vitro data also indicate that APOBEC3G and APOBEC3F are partially resistant to vif (3), suggesting a more fundamental role for these APOBEC3 proteins in directing HIV-1 genetic variation. However, at present little is known of the disease-modulating effects of APOBEC3G and/or APOBEC3F in HIV-infected patients, and it is uncertain if APOBEC3-mediated HIV DNA editing in vivo requires permissive conditions, such as defective vif activity and/or APOBEC3 genetic variation. Here, we have utilized near-full-length clade B HIV-1 proviral DNA sequences (an average of 6,820 ± 1,187 nucleotides/subject; total, 11,202 G→A substitutions) from 127 HIV-infected, antiretroviral therapy-naïve individuals to address each of these issues at a population level.
MATERIALS AND METHODS
Patient selection.
To be included in the study, pretreatment proviral HIV-1 DNA sequences were required to be of clade B (see below) and of sufficient length (>1,000 nucleotides). These criteria allowed the inclusion of 136 adult HIV-infected patients from the Western Australian HIV cohort (19), representing a predominantly Caucasian (84%) male (88%) population who had acquired HIV infection through sexual contact (83%). All sequences were utilized in the construction of the population consensus HIV-1 sequence in this study.
For the analysis of HIV-1 hypermutation based on proportions of nonconsensus nucleotides representing G→A substitutions in one of the dinucleotide sequence contexts (GA, GG, GC, or GT), a further nine sequences with 77 or fewer nucleotide substitutions from the consensus were omitted. These formed a tight cluster of outliers disparate from the remainder of the sample and provided potentially unstable estimates of the relevant proportions which could corrupt results. Our analyses were thus based on 127 cases.
HIV-1 clade assignment.
HIV sequences were analyzed by phylogenetic analysis using Molecular Evolutionary Genetic Analysis, version 3 (MEGA 3.0; http://www.megasoftware.net) as follows. The nucleic acid sequences for the individual HIV-1 protein products (e.g., p17, p24, and reverse transcriptase) were extracted from the database. These were combined with 6 to 15 sequences from each of clades A, B, C, D, and F as well as the sequence for HXB2 and the M-group ancestral sequence; these reference sequences were downloaded from the Los Alamos HIV sequence database. Phylogenetic trees were constructed using the parameters (i) neighbor-joining tree inference, (ii) pairwise deletion, and (iii) the Kumara two-parameter substitution model. The resulting trees were then rooted on the M-group ancestral sequence and inspected. Most of the sequences clearly sorted with reference sequences of a particular clade, and these were assigned as belonging to that clade. Sequences that did not clearly belong to a specific clade were assigned as unknown. A final assignment of clade was made based on the assignments for all of the proteins. If all of the protein products for a particular patient were unambiguously assigned to the same clade, then that sample was assigned to that clade. If all protein products were of the same clade except for one protein product assigned as ambiguous, the consensus clade was again assigned to that sample. Patient samples in which the proteins belonged to several clades were noted as such (e.g., AB, BC) and excluded from the study.
Amplification and sequencing of HIV-1 proviral DNA, measurement of pretreatment HIV RNA levels, and HLA and CCR5 genotyping.
Amplification and sequencing of HIV-1 proviral DNA, measurement of pretreatment HIV RNA levels, and HLA and CCR5 genotyping were performed as previously described in the work of Moore et al. (23). Patient DNA was extracted from blood samples by use of a QIAGEN DNA extraction kit according to the manufacturer's instructions. The PCR conditions and primers used for the full-length amplification of the proviral HIV genome have been previously described (26). Briefly, the first-round PCR was performed with an Expand long-template PCR kit (Boehringer Mannheim) to produce a 9-kb amplified product. This first round product was then used as a template for 13 individual nested PCRs using Taq polymerase (Boehringer Mannheim) to amplify the entire HIV genome from gag p17 to the 3′ long terminal repeat. First- and second-round PCRs were performed on ABI 9700 and 9600 thermocyclers. Successfully amplified PCR products were sequenced in the reverse and forward directions using BigDye Terminator ready reaction prism kits (v3). The samples were electrophoresed on an ABI 3100 genetic analyzer, and sequencing data analyzed using ABI software package Seqscape Version 1.1. Mixtures (sites where more than one nucleotide was observed) were named according to the IUPAC standard. Sites that were unable to be assigned were designated as “N” and excluded from the analyses. Nucleotide sequence length within the study population averaged 6,820 ± 1,187 nucleotides (range, 1,329 to 8,768 nucleotides).
Analysis of G→A substitutions.
To estimate G→A substitutions, individual HIV-1 proviral DNA sequences were aligned against the population consensus clade B sequence (n = 136). Only G→A substitutions where the nucleotide at position +1 in the sample matched the corresponding nucleotide in the consensus sequence were examined (where GA, GC, GG, and GT dinucleotides in the consensus sequence were observed as AA, AC, AG, and AT, respectively, in the sample sequence). Mixture nucleotide results obtained from chromatograph analysis (indicating the presence of mixed nucleotide populations) were assigned as missing values. To estimate “general” G→A hypermutation for each sequence (i.e., without reference to the expected dinucleotide sequence context for APOBEC3F or APOBEC3G), we incorporated two previously used measures of hypermutation (27), G→A preference (#G→A substitutions/#all mutations, where # indicates “number of”) and G→A burden (#G→A substitutions/ #consensus G) into a single formula:
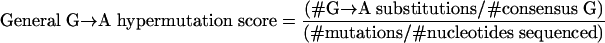 |
This represents the proportion of all mutations that are G→A substitutions adjusted for the proportion of nucleotides sequenced that are G in the consensus sequence.
Analysis of APOBEC3G and APOBEC3F target motifs.
To investigate the contribution of APOBEC3G (3G) and APOBEC3F (3F) to hypermutation, we examined the dinucleotide sequence contexts of G→A substitutions using the following formulae:
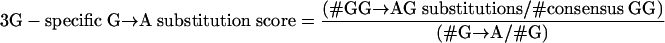 |
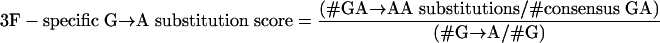 |
These represent the proportion of all G→A substitutions that occurred in the GG (APOBEC3G) and GA (APOBEC3F) contexts adjusted for the number of available GG (APOBEC3G) and GA (APOBEC3F) dinucleotides.
Analysis of APOBEC3G-mediated hypermutation (HM-3G) and HM-3F.
We combined the hypermutation formula with either the 3G or the 3F formula to identify sequences that had both an inordinate number of G→A substitutions relative to other substitutions and a high preference for G→A substitutions specifically in the sequence context targeted by either APOBEC3G or APOBEC3F as follows:
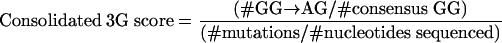 |
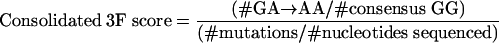 |
Thus, these represent the proportions of all mutations that were G→A substitutions that occurred in the GG (consolidated 3G score) and GA (consolidated 3F score) dinucleotide contexts, adjusted for nucleotide availability in the consensus sequence.
Histogram and Q-Q plots of the consolidated 3G values (log10 transformed) suggested a bimodal distribution with a normally distributed main group and a smaller group with higher values. The bimodal distribution was fitted by a mixture of two normal distributions using maximum likelihood, giving estimated distributions with means ± standard deviations (SD) of −0.277 ± 0.151 and 0.417 ± 0.151, respectively, with an estimated 9.0% of observations in the latter group. The presence of a mixture was highly significant (P < 0.00005; likelihood ratio test). The likelihood of belonging to the upper group was higher for the largest 12 observations (9.4%), with one likelihood ratio of 8 and the rest greater than 140. Hierarchical cluster analysis using between-group and centroid linkage both gave two clear groups with numbers of 115 and 12, as did k-means clustering. The nonhypermutated (NH) group is approximately normal. Corresponding cluster analyses of the non-APOBEC3G-hypermutated cases suggests three cases with inordinate consolidated 3F values.
APOBEC3G allele frequencies.
From eight participants with the highest G→A hypermutation scores and a pooled DNA control sample (n = 187 Caucasian individuals) (32), the entire APOBEC3G gene, including 2 kb 5′ of the transcription start site and 1.0 kb 3′ of the 3′ untranslated region, was amplified as two products of approximately 6.5 kb (3′ half) and 8 kb (5′ half) in size. For amplification of each product, 1 μl of DNA was amplified in a volume of 50 μl containing 10× Hi-Fi PCR buffer, 2 mM MgSO4 (1.5 mM MgSO4 for the 3′ half), 0.8 mM deoxynucleoside triphosphate mix, 100 nM of each forward and reverse primer, 1 U of PlatinumTaq Hi-Fi, and 2% dimethyl sulfoxide (1.5% for the 5′ half) under the following conditions: 94°C for 30 s, followed by 35 cycles of 94°C for 30 s, 63°C for 30 s (60°C for the 5′ half), and then by a 9-min extension step at 68°C. The PCR products were then purified using Exosap according to the manufacturer's protocol and sequenced using overlapping primers (see Table S1 in the supplemental material) and a BigDye Terminator kit, version 3.3 (PE, Applied Biosystems). The allele frequencies of the pooled DNA sample were determined by chromatograph relative peak height and compared to the allele frequencies for the eight patients harboring the HIV-1 proviral sequence with the greatest G→A hypermutation scores.
Statistical analysis.
Statistical analyses were carried out using SPSS software (version 12.0.1; SPSS Inc.). All values are expressed as means ± standard deviations unless otherwise stated. Where data were normally distributed, analysis of variance was used for group comparisons, with correction for multiple comparisons utilizing the Bonferroni method where appropriate. For nonnormal data, Mann-Whitney tests were used. The presence of in-frame stop codons in vif amino acid sequences was determined by putative translation of the proviral DNA sequences. With reference to analyses of the determinants of plasma HIV RNA levels prior to antiretroviral therapy, HLA-B27 (n = 7) and HLA-B58 (n = 4) were also considered as covariates along with HLA-B57 (n = 12) and the CCR5Δ32 genotype (n = 25).
RESULTS
Analysis of G→A substitutions.
We first sought to ascertain the population distribution of HIV-1 G→A substitutions in order to identify hypermutated HIV-1 sequences within the context of natural in vivo sequence variation. The study was restricted to sequences identified as clade B by phylogenetic analysis (n = 127), involving a study population of predominantly male (87%) Caucasian (84%) patients with chronic HIV-1 infection. All participants were antiretroviral therapy naïve at the time of clinical assessment and collection of proviral HIV-1 DNA for sequencing, as reflected in the distribution of HIV RNA/ml viral load (mean ± SD, 4.903 ± 0.760 log10 copies), CD4+ T cell counts (380.6 ± 298.3 cells/ml), and percentage of CD4+ T cells (19.3% ± 10.9%). To investigate G→A substitutions at the population level, we aligned each individual's HIV-1 proviral sequence to the population consensus sequence and for each sequence considered two measures of hypermutation as previously described (27): (i) the preference for G→A substitutions relative to all other substitutions (defined by the proportion of all mutations that were G→A substitutions) and (ii) the burden of G→A substitutions relative to the number of available consensus guanine nucleotides (defined by the proportion of consensus guanines that exhibited G→A substitutions). As expected, we found that G→A substitutions were the most common substitution observed (median ± interquartile range, 21.0% ± 4.0% of all substitutions), and there was a highly significant correlation between G→A preference and G→A burden in the study population (Spearman's r = 0.631; P < 0.001), as shown in Fig. 1A.
FIG. 1.

G→A substitutions in HIV-1 proviral DNA with inordinate G→A preference and G→A burden occur predominantly in the dinucleotide sequence contexts targeted by APOBEC3G (GG) and, to a lesser extent, in those targeted by APOBEC3F (GA). (A) G→A preference (#G→A substitutions/all mutations) was highly significantly correlated to G→A burden (#G→ substitutions/#consensus G) (r = 0.853; P < 0.001). (B and C) The general G→A hypermutation scores were significantly correlated with the 3G-specific G→A substitution scores (r = 0.318; P < 0.001) but not with the 3F-specific G→A substitution scores (r = 0.065; P = 0.465). Red shaded circles, HM-3G sequences determined by mixed population analysis of the consolidated 3G score; black open triangles, HM-3F sequences determined by mixed population analysis of the consolidated 3F score; blue open circles, NH sequences determined by mixed population analyses of the consolidated 3G and consolidated 3F scores; †, general G→A hypermutation score (#G→A substitutions/#consensus G)/(#mutations/#nucleotides sequenced); *, 3G-specific G→A substitution score (#GG→AG substitutions/#consensus GG)/(#G→A/#G); ‡, 3F-specific G→A substitution score (#GA→AA substitutions/#consensus GA)/(#G→A/#G).
We then incorporated both G→A burden and G→A preference into a single index (general G→A hypermutation score; see Materials and Methods) to ascertain if the distribution of G→A substitutions conformed to a model in which a proportion of sequences could be described as hypermutated relative to natural sequence variation. Utilizing analyses of population mixtures, these data were best described by a bimodal mixed distribution rather than as data from a single population (P < 0.001), consistent with the hypothesis that a proportion of proviral DNA sequences exhibited HIV-1 hypermutation.
Analysis of APOBEC3G and APOBEC3F dinucleotide target motifs.
As mentioned previously, APOBEC3G and APOBEC3F are known to target specific single-stranded polynucleotide DNA motifs. Therefore, in order to further characterize and investigate APOBEC3-associated HIV-1 hypermutation, we further examined the preference for G→A substitutions in the APOBEC3G (GG) and APOBEC3F (GA) dinucleotide contexts. This was first accomplished by investigating the dinucleotide sequence context for G→A substitutions (3G- and 3F-specific G→A substitution scores; see Materials and Methods). Again, the population distribution of the 3G-specific G→A substitution scores indicated the presence of APOBEC3G-mediated G→A substitutions in a proportion of sequences, in that there were two distinct clusters in the distribution of GG→AG substitutions in the study population (P < 0.001) (Fig. 1B). In contrast, while there were three relatively extreme 3F-specific G→A substitution scores, it was not possible to demonstrate clear clusters (Fig. 1C).
Classification of APOBEC3G- and APOBEC3F-hypermutated HIV-1 sequences.
In order to formally identify sequences with evidence of HM-3G, we then analyzed the population distribution of HIV-1 G→A substitutions incorporating both (i) G→A preference relative to other mutations and (ii) preference for G→A substitutions within the GG context (consolidated 3G score; see Materials and Methods) (Fig. 2). Mixture models and cluster analyses consistently identified 12 sequences (9.4%) that fulfilled these criteria (P < 0.0005). As shown in Table 1, these sequences exhibited a 1.9-fold preference for G→A substitutions on average and a 3.1-fold increase in G→A burden compared with the remaining NH sequences. In addition, 42.7% ± 8.7% of all G→A substitutions were in the GG context, compared with 17.7% ± 5.0% in the NH sequences. The average proportion of consensus guanine bases that demonstrated G→A substitutions in these hypermutated sequences (14.2%) was consistent with previous descriptions of hypermutated HIV-1 proviral DNA obtained from env gene sequences in vivo (16%) (15) and utilizing Δvif virions in vitro (24).
FIG. 2.

The consolidated 3G scores reflect a bimodal distribution. The bimodal distribution of the consolidated 3G scores [(#GG→GA/#consensus GG)/(#mutations/#nucleotides sequenced)] was fitted by a mixture of two normal distributions utilizing maximum likelihood, giving estimated distributions with means ± SD of −0.277 ± 0.151 (blue shaded bars; subsequently defined as NH sequences) and 0.417 ± 0.151 (red partially shaded bars; subsequently defined as HM-3G sequences), respectively. The presence of a population mixture was highly significant (P < 0.0005; likelihood ratio test).
TABLE 1.
Patient and sequence characteristics of patients harboring putative APOBEC3G-hypermutated, APOBEC3F-hypermutated, and nonhypermutated HIV-1 proviral DNA
| Characteristic or score | Value for indicated type of DNAa
|
||
|---|---|---|---|
| NH | HM-3G | HM-3F | |
| Patient characteristics | |||
| No. | 112 | 12 | 3 |
| CD4+ count (cells/ml) | 367.2 ± 306.1 | 491.3 ± 236.7 | 421.3 ± 152.1 |
| CD4+ percentage | 18.5 ± 10.7 | 24.7 ± 11.4 | 28.3 ± 8.3 |
| Viral load (log10 copies/ml) | 4.981 ± 0.747 | 4.476 ± 0.50* | 3.721 ± 0.680* |
| HIV-1 sequence characteristics | |||
| No. of nucleotides sequenced per patient | 6,906 ± 1,016 | 6,571 ± 1,549 | 4,591 ± 3,182 |
| No. of non-G→A mutations | 290 ± 66 | 295 ± 97 | 246 ± 129 |
| No. of G→A substitutions | 75 ± 18 | 210 ± 107 | 110 ± 40 |
| G→A rate (per 100 nucleotides) | 1.08 ± 0.21 | 3.4 ± 1.8 | 3.3 ± 2.4 |
| G→A substitutions: all substitutions | 0.206 ± 0.029 | 0.40 ± 0.123 | 0.321 ± 0.049 |
| G→A burden (% of consensus G's) | 4.6 ± 0.9 | 14.2 ± 7.5 | 14.9 ± 11.4 |
| GG (% of G→A substitutions)b | 17.7 ± 5.0 | 42.7 ± 8.7 | 10.8 ± 5.2 |
| GA (% of G→A substitutions)b | 32.1 ± 6.0 | 26.1 ± 7.0 | 59.9 ± 11.3 |
| Hypermutation scoresc | |||
| General G→A hypermutation | −0.06 ± 0.06 | 0.22 ± 0.11 | 0.14 ± 0.08 |
| 3G-specific G→A substitutions | −0.21 ± 0.13 | 0.19 ± 0.11 | −0.45 ± 0.25 |
| 3F-specific G→A substitutions | −0.05 ± 0.08 | −0.15 ± 0.11 | 0.22 ± 0.06 |
| Consolidated 3G | −0.28 ± 0.15 | 0.41 ± 0.16 | −0.30 ± 0.16 |
| Consolidated 3F | −0.12 ± 0.11 | 0.07 ± 0.15 | 0.36 ± 0.15 |
Numbers are expressed as means ± standard deviations where appropriate. *, <5% significance level compared to NH sequences.
GG and GA represent percentages of G→A substitutions that occurred in the GG and GA dinucleotide contexts, respectively.
See Materials and Methods for log10 transformed values; significance not determined for HIV-1 sequence characteristics or hypermutation scores, as these were the basis of the classifications.
Three additional sequences showed a strong preference for HM-3F using these criteria (Table 1), with 59.9% ± 11.3% of all G→A substitutions occurring in the GA context, compared with only 26.1% ± 7.0% and 32.1% ± 6.0% in the HM-3G and NH sequences, respectively.
While G→A substitutions were the most common substitution observed for the 112 NH sequences, there was no association between the general G→A hypermutation score and the 3G-specific G→A substitution score (Pearson's r = 0.093; P = 0.331) or the 3F-specific G→A substitution score (r = 0.140; P = 0.142). Hence, it is unlikely that APOBEC3G- or APOBEC3F-mediated DNA editing contributes significantly to the rate of G→A substitutions (identified by bulk PCR sequencing) within these nonhypermutated proviral DNA sequences.
HIV-1 genomic distribution of G→A hypermutation.
From the data presented in Fig. 3, measures of general G→A hypermutation (Fig. 3A), APOBEC3G-specific G→A substitutions (Fig. 3B), and APOBEC3G-mediated hypermutation (Fig. 3C) were distributed relatively evenly along the genome. Consistent with results from Yu and colleagues (41), general G→A hypermutation and APOBEC3G-mediated hypermutation scores in HM-3G sequences were significantly higher for pol (P < 0.001) than for gag (P = 0.005); in contrast, however, they were significantly lower for env than for pol (P values for both were <0.001). Furthermore, consistent with results from Wurtzer and colleagues (37), the distance from the central polypurine tract was significantly negatively correlated to the general G→A hypermutation (r = −0.283; P = 0.007), 3G-specific G→A substitution (r = −0.250; P = 0.020), and consolidated 3G (r = −0.297; P = 0.006) scores.
FIG. 3.

General G→A hypermutation [(#G→A substitutions/#consensus G)/(#mutations/#nucleotides sequenced)], 3G-specific G→A substitutions [(#GG→AG substitutions/#consensus GG)/(#G→A/#G)], and consolidated 3G [(#GA→AA substitutions/#consensus GA)/(#G→A/#G)] scores were relatively constant along the HIV-1 genome. Red shaded circles, HM-3G sequences; blue open circles, NH sequences; x axis, nucleotide position relative to HXB2 (GenBank accession number K03455). y axis values are log10 transformed; curves are locally weighted (Loess) smoothers.
Association of vif amino acid polymorphisms and G→A hypermutation.
We subsequently sought to investigate associations between vif amino acid polymorphisms and hypermutation. For such analyses, we performed a putative translation of available vif sequences (n = 124) and examined vif polymorphism within the NH sequences (vif amino acids 1 to 193) compared with the corresponding amino acid in the HM-3G sequences. HM-3F sequences were omitted from these analyses so that specific associations relevant to the APOBEC3G-vif interaction could be examined, as it has recently been reported that specific vif amino acid polymorphisms differentially influence APOBEC3G versus APOBEC3F interaction (31).
vif peptide sequences derived from the HM-3G group were significantly different from NH vif sequences in terms of a higher overall rate of nonconsensus amino acids (P < 0.0005; Mantel-Haenszel). The strongest associations were tryptophan-to-in-frame stop substitutions at positions 70, 89, 174, and 21 and substitution of isoleucine for the methionine start codon (all Pc [corrected P] values were <0.04 by the Bonferroni method). These substitutions, which would be anticipated to code for truncated and functionally defective vif proteins, are all in the trinucleotide sequence context targeted by APOBEC3G (TGG) and therefore are likely to have resulted from, rather than caused, HIV-1 hypermutation. HM-3G sequences were also associated with an R90K polymorphism (Pc = 0.07; Bonferroni); again, however, this could be attributed to the action of APOBEC3G. No other vif polymorphisms were found to be significantly associated with APOBEC3G-associated hypermutation (all Pc values were >0.4).
Regarding the potential role of truncated vif sequences in permitting APOBEC3G-mediated HIV-1 hypermutation, it is notable that while HM-3G sequences were significantly associated with in-frame stop codons specifically in vif (P < 0.001), in-frame stop codons in non-vif or non-vif, non-env proteins were not associated with HM-3G sequences (P values were 0.20 and 0.63, respectively). Additionally, HIV-1 hypermutation was not evident in the two sequences in which the only vif in-frame stop codon was at vif amino acid position 153 (leucine) or 174 (tryptophan). In these sequences, G→A substitutions accounted for only 20.7% and 24.9% of all substitutions, affecting only 4.0% and 5.0% of consensus guanine residues, respectively. It has previously been demonstrated that a naturally occurring vif protein truncated at tryptophan174 is indistinguishable from wild-type vif in terms of the maintenance of viral infectivity (25). Here, the occurrence of a naturally occurring vif protein truncated at leucine153 also suggests that the C-terminal 39 amino acids are dispensable for this protein's roles in promoting viral infectivity as well as counteracting APOBEC3G. Interestingly, despite the codon for tryptophan being a target for APOBEC3G, tryptophan residues 5, 11, and 79 were entirely conserved in the hypermutated vif sequences and are therefore unlikely to play a role in inhibiting APOBEC3G or APOBEC3F activity, in contrast to data from Tian and colleagues (33).
We also identified vif polymorphisms that were unique to the HM-3G sequences within this study population, given previous evidence that sequence variation at a number of critical residues can affect vif-APOBEC3G interactions (29, 35). Twenty-three vif amino acid variants were found uniquely in HM-3G sequences (see Fig. 5), of which 82% were within regions previously shown to be required for vif-APOBEC3G interaction (29, 35). Of these, 13 could not have resulted from an APOBEC3G-mediated G→A substitution of the NH consensus amino acid, all of which were located within the vif N terminus. It is also interesting that a charged consensus amino acid was present in 14/23 of these positions and that the variant vif sequence was frequently associated with either a reversal of the charge state (6/14, 43%) or substitution of a neutral amino acid (3/14, 21%). An additional three vif amino acids were unique to the two HM-3F vif sequences available and also could not have resulted from a G→A substitution of any of the alternative amino acids present in the NH sequences. Again, two of the HM-3F unique amino acids were located within the vif N terminus (L98 and S103).
FIG. 5.

Schematic representation of HM and truncated vif proteins identified in vivo from putative translation of proviral DNA sequences. Shaded bars indicate translated vif sequences, unshaded bars indicate untranslated vif sequences, and partially shaded bars indicate regions not sequenced. Red lines indicate unique polymorphisms from consensus that could not be attributed to the corresponding APOBEC3 enzyme, and blue lines indicate unique polymorphisms from consensus that could potentially be attributed to the corresponding APOBEC3 enzyme. Sequences are ordered by HM-3G, HM-3F, and NH in descending order according to the consolidated 3G score. Amino acids below the peptides indicate consensus, and amino acids above the peptides indicate amino acids unique to the individual peptide sequence.
Figure 4 demonstrates the high degree of vif polymorphism that appears to be tolerated without apparently increasing viral susceptibility to the effects of APOBEC3G and/or APOBEC3F. Within the nonhypermutated group, polymorphism was observed at the serine144 and leucine148 residues (underlined) of the SOCS-box N-terminal BC-box motif (144SLQYLA149) required for vif phosphorylation (40) and Elongin BC and Cul5 binding (22), respectively, and at the proline162 (1.0%) residue of the SOCS-box C-terminal motif 161PPLP164, required for vif multimerization and subsequent vif function (39). In contrast, the existence of polymorphism at these sites in our population suggests that they may not be essential for maintaining vif function. However, the recently described Hx5Cx17-18Cx3-5H zinc-binding motif (38), critical for assembly and activity of the vif-Cul5-E3 ligase (18), was entirely conserved.
FIG. 4.

The consensus vif amino acid sequence of the nonhypermutated sequences. The consensus sequence of the nonhypermutated sequences shown here was identical to the consensus vif sequence of the hypermutated HIV-1 sequences. To determine the consensus amino acid sequence, individual sequences were putatively translated, and the most common amino acid at each location was deemed the consensus. Blue bars indicate frequencies of nonconsensus amino acids. Underlined letters indicate previously described motifs, with open bars indicating the frequency of nonconsensus amino acids at these sites. See the text for references regarding vif regulatory motifs. Numbers indicate vif amino acid numbers. *, stop codon; 1, Fujita et al., 2004 (reference 8); 2, Mehle et al., 2004 (reference 22); 3, Yang et al., 2003 (reference 39); 4, Ochsenbauer et al., 1996 (reference 25).
APOBEC3G genetic variation and G→A hypermutation.
To assess the contribution of APOBEC3G genetic variation to hypermutation, we sequenced the entire 10.5-kb APOBEC3G gene, including 2 kb 5′ of the transcription start site and 0.5 kb 3′ of the 3′ untranslated region. We initially compared the frequency of APOBEC3G alleles from eight patients with the greatest G→A burden to that estimated from a pooled DNA sample (187 Caucasian individuals) (32) as a control, based on relative chromatogram peak height. As demonstrated in Table 2, the APOBEC3G amino acid sequence was highly conserved in this predominantly Caucasian study population. Of the 22 single nucleotide polymorphisms (SNPs) identified (17 previously described and 5 novel SNPs), significant differences in allele frequencies between the patients with hypermutated sequences and the pooled DNA control sample were evident at positions 625 (9607609) and 6,892 (5757467) relative to the transcription start site (Table 2). We then genotyped the 625 and 6,892 SNPs for patients that had DNA available (n = 119). The frequency of the 6,892 C allele tended to be higher in patients with evidence of APOBEC3G-mediated hypermutation (50.0% versus 29.9%; P = 0.062), with homozygosity for the 6,892 C allele present in 25% of patients with evidence of APOBEC3G-mediated hypermutation compared with 7.5% of patients with nonhypermutated sequences (P = 0.082). However, this marginally significant univariate association was abrogated after adjusting for multiple comparisons (Pc > 0.5). The frequencies of the 625 C allele were similar for patients with evidence of APOBEC3G-mediated hypermutation and for patients harboring NH sequences (allele frequency, 75.0% versus 77.2% [P = 0.80]; CC genotype frequency, 58.3% versus 57.3% [P = 1.0]).
TABLE 2.
APOBEC3G allele frequencies among eight patients with marked G→A hypermutationa
| NCBI dbSNP IDb | Positionc | Frequency ofd:
|
No. of HM-3G patients that weree:
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Control sample
|
Allele 2 from the work of Do et al. (7) | HM-3G patient
|
A1.A1 | A1.A2 | A2.A2 | ||||
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | ||||||
| 7291971 | −1,193 | 0.60 | 0.40 | 0.40 | 0.44 | 0.56 | 2 | 3 | 3 |
| 6519166 | −1,190 | 0.70 | 0.30 | 0.31 | 0.56 | 0.44 | 2 | 5 | 1 |
| 5750743 | −321 | 0.50 | 0.50 | 0.28 | 0.44 | 0.56 | 1 | 5 | 2 |
| 12160242 | 480 | >0.90 | <0.10 | 0.09 | 0.94 | 0.06 | 7 | 1 | 0 |
| 9607609 | 625 | 0.80 | 0.20 | ND | 0.38 | 0.62 | 2 | 2 | 4 |
| 5757465 | 3,756† | 0.60 | 0.40 | 0.51 | 0.75 | 0.25 | 4 | 4 | 0 |
| 12158985 | 5,985 | >0.90 | <0.10 | 0.07 | 0.88 | 0.12 | 6 | 2 | 0 |
| 2294366 | 5,986 | 0.70 | 0.30 | 0.34 | 0.56 | 0.44 | 2 | 5 | 1 |
| 2294367 | 6,207 | 0.60 | 0.40 | 0.42 | 0.44 | 0.56 | 2 | 3 | 3 |
| NL | 6,848 | >0.90 | <0.10 | ND | 0.88 | 0.12 | 6 | 2 | 0 |
| 3091374 | 6,878 | 0.50 | 0.50 | ND | 0.44 | 0.56 | 2 | 3 | 3 |
| 5757467 | 6,892 | 0.70 | 0.30 | ND | 0.31 | 0.69 | 0 | 5 | 3 |
| 5757468 | 6,893 | 0.70 | 0.30 | ND | 0.56 | 0.44 | 2 | 5 | 1 |
| NL | 6,894 | >0.90 | <0.10 | ND | 0.88 | 0.12 | 6 | 2 | 0 |
| NL | 8,984‡ | >0.90 | <0.10 | ND | 0.88 | 0.12 | 6 | 2 | 0 |
| 5757471 | 9,218 | >0.90 | <0.10 | ND | 0.94 | 0.06 | 7 | 1 | 0 |
| 28474760 | 9,250 | >0.90 | <0.10 | 0.07 | 0.88 | 0.12 | 6 | 2 | 0 |
| 28474761 | 9,432 | >0.90 | <0.10 | 0.12 | 0.88 | 0.12 | 6 | 2 | 0 |
| NL | 10,393 | 0.50 | 0.50 | ND | 0.92 | 0.08 | 5 | 1 | 0 |
| NL | 10,399 | 0.70 | 0.30 | ND | 1.00 | 0.0 | 8 | 0 | 0 |
| 5757472 | 10,600 | 0.50 | 0.50 | ND | 0.44 | 0.56 | 1 | 5 | 2 |
| 3891126 | 10,811 | >0.90 | <0.10 | ND | 0.88 | 0.12 | 6 | 2 | 0 |
Comparisons with a pooled DNA control sample and with results from a published study are shown. Results in boldface are those for which significant differences in allele frequencies between patients with hypermutated sequences and the pooled DNA control sample were evident.
NCBI dbSNP ID, National Center for Biotechnology Information SNP database reference number. NL, novel SNP.
Positions are relative to the transcription start site with reference to NCBI accession number NT_011520. †, synonymous SNP; ‡, nonsynonymous SNP.
Allele frequencies of the pooled DNA sample were estimated by chromatogram relative peak height. Significant differences in the allele frequencies of the 625 and 6,892 SNPs were evident between the hypermutated patients and the pooled DNA sample. A result of <0.10 indicates that allele frequency was below the limit of detection. ND, not determined.
A1.A1, homozygous for allele 1; A1.A2, heterozygous; A2.A2, homozygous for allele 2.
G→A hypermutation and HIV-1 viremia.
The biological and clinical relevance of HIV-1 hypermutation in proviral DNA sequences is ultimately a function of its impact on productive HIV infection, measured by the level of viremia in plasma samples. We therefore sought to investigate the effect of HIV-1 hypermutation on pretreatment viremia in this study population. Univariate analysis indicates that patients harboring hypermutated sequence as defined had viral loads significantly lower than those with nonhypermutated sequence (4.32 ± 0.60 versus 4.98 ± 0.75 log10copies HIV RNA/ml; P = 0.001). By use of linear regression models, this association between hypermutation and lower pretreatment viremia remained highly significant (P = 0.013) even after adjusting for CD4+ percentage (P < 0.001) and presence of at least one of the known protective host alleles, CCR5Δ32 (17), HLA-B57, HLA-B58, or HLA-B27 (9) (P = 0.096), and approximates 67% and 40% reductions in viral loads attributable to hypermutation and host protective alleles, respectively. Hence, the clinical influence of HIV-1 hypermutation appears to be demonstrable within this group of patients, who manifest a marked preference for G→A substitutions in an APOBEC3G or APOBEC3F sequence context.
DISCUSSION
The data presented in this population-based study of clade B HIV-1 sequences suggest that HIV-1 G→A hypermutation is a prevalent phenomenon associated with significantly reduced plasma HIV RNA levels in vivo, indicating that APOBEC3G- and APOBEC3F-mediated hypermutation can take its place alongside other protective host genetic factors as a clinically and biologically relevant antiretroviral restriction factor. Indeed, the reduced HIV-1 viremia associated with hypermutation was substantially greater than that exerted by known host factors, such as the CCR5Δ32 chemokine receptor variant, and remained highly significant after adjusting for these variables as well as for the influence of CD4+ count. Such reductions in viral load are substantially greater than those previously associated with protective HLA alleles (28) and are comparable to the effects of zidovudine monotherapy in early studies of antiretroviral treatment (4).
Here, it was estimated that hypermutated proviral DNA was present in 12% of study participants by use of a bulk PCR sequencing approach that approximates the dominant species within a heterogeneous viral population. In many respects, these findings complement the work of Kieffer and colleagues (14), who examined the phenomenon of in vivo hypermutation in detail utilizing 319 pol clones derived from nine patient samples. This study revealed at least one hypermutated proviral DNA sequence from each individual examined, although hypermutated sequences accounted for less than 10% of the proviral population within an individual. Similar results have been obtained by Janini et al. (11), who detected the presence of G→A hypermutation in 45% of HIV protease sequences from a Tanzanian study population. These observations are consistent with in vitro data demonstrating low-level APOBEC3G- and APOBEC3F-mediated cytidine deaminase activity directed against HIV-1 sequences even in the presence of functional vif (13, 21). It has also been suggested that the rapid turnover of vif through proteasomal degradation contributes to constitutively low cellular protein expression (34), thereby providing an incomplete barrier to APOBEC3G and/or APOBEC3F activity. In this study, we now provide evidence that hypermutated proviral HIV-1 DNA sequences can be demonstrated by bulk PCR methods in a significant minority of clade B HIV-1-infected individuals. Taken together, these data suggest that APOBEC3-mediated cytidine deamination is highly prevalent but effectively constrained by functional interactions with vif, so that G→A hypermutation is generally confined to a minor proportion of the overall viral population.
It should be noted that the sequence context targeted by APOBEC3G appears to specifically select for loss-of-function mutations, including within the vif genomic sequence. For example, targeted substitutions involving tryptophan residues (TGG) produce in-frame stop codons (TAG), while the vif start codon and adjacent aspartic acid residue also create an APOBEC3G target motif (ATGG) that results in loss of the methionine initiation signal following G→A substitution. To some extent, this susceptibility appears to have been overcome by the utilization of multiple alternative start codons at methionine residues 8, 16, and 29, although it is notable that the capacity to counteract APOBEC3G activity appears to be lost as a consequence of these N-terminal truncations (31, 34). In this study, substitutions associated with truncated vif sequences could be entirely explained by APOBEC3G-mediated cytidine deamination (Fig. 5), although it is notable that hypermutation was associated specifically with vif in-frame stop codons but not with in-frame stop codons in non-vif viral peptides. Hence, while it is difficult to attribute causality in the relationship between hypermutation and defective vif, the most parsimonious explanation appears to be provided by a mechanism in which APOBEC3-mediated G→A substitutions, occurring in a relatively nonpermissive cellular environment but targeting TGG trinucleotide motifs stochastically along the genome, can become unconstrained if this process selects for defective vif sequences.
Although the translated vif sequences in this cohort were highly polymorphic (Fig. 4), there were 23 vif amino acid variants unique to HIV-1 hypermutated sequences, of which 13 associated with HM-3G sequences and three unique to HM-3F sequences could not have resulted from cytidine deamination by the relevant APOBEC3 protein (Fig. 5). These data are consistent with those that suggest the vif N terminus contains distinct motifs that are likely to interface with APOBEC3G and APOBEC3F (33, 35) and may therefore represent true vif allelic variants that facilitate hypermutation. Although the associated amino acids differed, four of the twenty-three vif amino acids unique to HM-3G sequences (K45, E75, E138, and K185) occurred at positions previously associated with naturally occurring nonfunctional vif variants (31).
With reference to the nonhypermutated vif sequences, we observed polymorphism at residues within key motifs shown to be required for vif phosphorylation (40), vif-E3 ligase complex formation (22), vif multimerization (39), and β-sheet formation (8). In this study, however, these variants were not associated with APOBEC3-mediated hypermutation, suggesting that polymorphisms within these motifs are tolerated without significantly compromising vif function. While it is unclear if these polymorphisms alter the efficiency of polyubiquitination and proteasomal degradation of the vif-APOBEC3G-E3 ligase complex in vivo, the lack of hypermutation in these sequences could potentially be attributed to an undiminished capacity for vif-APOBEC3G complex formation, which is sufficient to restrict APOBEC3G-mediated hypermutation in vitro (13). Despite the high degree of vif polymorphism, several stretches of conserved residues, most notably the first 18 N-terminal amino acids, were observed and may represent novel drug targets.
The dinucleotide sequence context of the G→A substitutions suggests that APOBEC3G was the dominant contributor to hypermutation in this study group. These findings are consistent with the previously mentioned in vivo study by Kieffer et al., which involved clade B HIV-1 sequences (14). In contrast, two studies involving Tanzanian study populations infected with predominantly non-clade B virus (11, 15) indicate less bias towards the APOBEC3G context. These differences warrant further exploration, particularly as a nonsynonymous APOBEC3G 186R polymorphism associated with accelerated progression to AIDS (and therefore implying loss of APOBEC3G function) has been found to be frequent in African Americans (37%) but rare in European Americans (5%) (1). Hence, a relatively greater contribution of APOBEC3F to HIV-1 cytidine deaminase editing may be anticipated in at least some populations of African origin. In this study, there was no convincing evidence that APOBEC3G genetic variation contributed to the development of HIV-1 hypermutation, and it is notable that we were unable to identify significant amino acid variation within APOBEC3G sequences in these Caucasian populations (Table 2). A marginal univariate association between an intronic APOBEC3G 6,892 C allele and hypermutation was demonstrated, although the role of this intronic polymorphism on APOBEC3G function is unclear. However it is possible that functional polymorphisms are located in the as-yet-undefined promoter or regulatory regions of this genetic locus, and in this regard it is notable that APOBEC3G mRNA levels in stimulated peripheral blood mononuclear cells have recently been found to exhibit an inverse correlation with HIV-1 pretreatment viremia (12), though no correlation exists in resting T cells (5).
In conclusion, this study provides strong support for the proposition that APOBEC3G-mediated cytosine deamination provides a potent source of innate HIV-1 restriction at the population level. Moreover, the prevalence and antiretroviral impact of APOBEC3G- and APOBEC3F-mediated HIV-1 hypermutation within the study population can be compared favorably with known host protective factors. Since Sheehy and colleagues first isolated the APOBEC3G gene 4 years ago and tentatively proposed a cytidine deaminase function for its product (30), considerable advances in the understanding of APOBEC3 proteins and their antiviral activity have been made. We believe that these data contribute to the premise that APOBEC3G comprises a form of innate antiviral resistance that is clinically and biologically relevant and may lend a fresh perspective to the area of HIV/AIDS therapies (2).
Supplementary Material
Acknowledgments
We declare that we have no competing financial interests. S. Gaudieri was supported by a Healy Fellowship from the Raine Medical Research Foundation.
We are indebted to all participants in the Western Australian HIV Cohort Study and to past and present laboratory staff of the Department of Clinical Immunology & Biochemical Genetics, Royal Perth Hospital, Western Australia, and the Centre for Clinical Immunology & Biomedical Statistics. In particular, we acknowledge the contribution of Filipa Carvalho in the area of HIV genomic sequencing. We thank Graeme Stewart for the kind donation of the pooled DNA sample, L. Park for HIV-1 clade assignment, and A. Rauch for critically reading the manuscript.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.An, P., G. Bleiber, P. Duggal, G. Nelson, M. May, B. Mangeat, I. Alobwede, D. Trono, D. Vlahov, S. Donfield, J. J. Goedert, J. Phair, S. Buchbinder, S. J. O'Brien, A. Telenti, and C. A. Winkler. 2004. APOBEC3G genetic variants and their influence on the progression to AIDS. J. Virol. 78:11070-11076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392-1396. [DOI] [PubMed] [Google Scholar]
- 3.Bishop, K. N., R. K. Holmes, A. M. Sheehy, and M. H. Malim. 2004. APOBEC-mediated editing of viral RNA. Science 305:645. [DOI] [PubMed] [Google Scholar]
- 4.Brun-Vezinet, F., C. Boucher, C. Loveday, D. Descamps, V. Fauveau, J. Izopet, D. Jeffries, S. Kaye, C. Krzyanowski, A. Nunn, R. Schuurman, J. M. Seigneurin, C. Tamalet, R. Tedder, J. Weber, G. J. Weverling, et al. 1997. HIV-1 viral load, phenotype, and resistance in a subset of drug-naive participants from the Delta trial. Lancet 350:983-990. [DOI] [PubMed] [Google Scholar]
- 5.Cho, S. J., H. Drechsler, R. C. Burke, M. Q. Arens, W. Powderly, and N. O. Davidson. 2006. APOBEC3F and APOBEC3G mRNA levels do not correlate with human immunodeficiency virus type 1 plasma viremia or CD4+ T-cell count. J. Virol. 80:2069-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conticello, S. G., R. S. Harris, and M. S. Neuberger. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009-2013. [DOI] [PubMed] [Google Scholar]
- 7.Do, H., A. Vasilescu, G. Diop, T. Hirtzig, S. C. Heath, C. Coulonges, J. Rappaport, A. Therwath, M. Lathrop, F. Matsuda, and J. F. Zagury. 2005. Exhaustive genotyping of the CEM15 (APOBEC3G) gene and absence of association with AIDS progression in a French cohort. J. Infect. Dis. 191:159-163. [DOI] [PubMed] [Google Scholar]
- 8.Fujita, M., H. Akari, A. Sakurai, A. Yoshida, T. Chiba, K. Tanaka, K. Strebel, and A. Adachi. 2004. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 6:791-798. [DOI] [PubMed] [Google Scholar]
- 9.Goulder, P. J., M. Bunce, P. Krausa, K. McIntyre, S. Crowley, B. Morgan, A. Edwards, P. Giangrande, R. E. Phillips, and A. J. McMichael. 1996. Novel, cross-restricted, conserved, and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV type 1 infection. AIDS Res. Hum. Retrovir. 12:1691-1698. [DOI] [PubMed] [Google Scholar]
- 10.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803-809. [DOI] [PubMed] [Google Scholar]
- 11.Janini, M., M. Rogers, D. R. Birx, and F. E. McCutchan. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T cells. J. Virol. 75:7973-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin, X., A. Brooks, H. Chen, R. Bennett, R. Reichman, and H. Smith. 2005. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J. Virol. 79:11513-11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kao, S., E. Miyagi, M. A. Khan, H. Takeuchi, S. Opi, R. Goila-Gaur, and K. Strebel. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kieffer, T. L., P. Kwon, R. E. Nettles, Y. Han, S. C. Ray, and R. F. Siliciano. 2005. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 79:1975-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koulinska, I. N., B. Chaplin, D. Mwakagile, M. Essex, and B. Renjifo. 2003. Hypermutation of HIV type 1 genomes isolated from infants soon after vertical infection. AIDS Res. Hum. Retrovir. 19:1115-1123. [DOI] [PubMed] [Google Scholar]
- 16.Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385-1391. [DOI] [PubMed] [Google Scholar]
- 17.Liu, R., W. A. Paxton, S. Choe, D. Ceradini, S. R. Martin, R. Horuk, M. E. MacDonald, H. Stuhlmann, R. A. Koup, and N. R. Landau. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86:367-377. [DOI] [PubMed] [Google Scholar]
- 18.Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 102:11444-11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallal, S. A. 1998. The Western Australian HIV Cohort Study, Perth, Australia. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 17(Suppl. 1):S23-S27. [DOI] [PubMed] [Google Scholar]
- 20.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99-103. [DOI] [PubMed] [Google Scholar]
- 21.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398-1403. [DOI] [PubMed] [Google Scholar]
- 22.Mehle, A., J. Goncalves, M. Santa-Marta, M. McPike, and D. Gabuzda. 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18:2861-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439-1443. [DOI] [PubMed] [Google Scholar]
- 24.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166-170. [DOI] [PubMed] [Google Scholar]
- 25.Ochsenbauer, C., V. Bosch, I. Oelze, and U. Wieland. 1996. Unimpaired function of a naturally occurring C terminally truncated vif gene product of human immunodeficiency virus type 1. J. Gen. Virol. 77:1389-1395. [DOI] [PubMed] [Google Scholar]
- 26.Oelrichs, R. B., V. A. Lawson, K. M. Coates, C. Chatfield, N. J. Deacon, and D. A. McPhee. 2000. Rapid full-length genomic sequencing of two cytopathically heterogeneous Australian primary HIV-1 isolates. J. Biomed. Sci. 7:128-135. [DOI] [PubMed] [Google Scholar]
- 27.Rose, P. P., and B. T. Korber. 2000. Detecting hypermutations in viral sequences with an emphasis on G→A hypermutation. Bioinformatics 16:400-401. [DOI] [PubMed] [Google Scholar]
- 28.Saah, A. J., D. R. Hoover, S. Weng, M. Carrington, J. Mellors, C. R. Rinaldo, Jr., D. Mann, R. Apple, J. P. Phair, R. Detels, S. O'Brien, C. Enger, P. Johnson, R. A. Kaslow, et al. 1998. Association of HLA profiles with early plasma viral load, CD4+ cell count and rate of progression to AIDS following acute HIV-1 infection. AIDS 12:2107-2113. [DOI] [PubMed] [Google Scholar]
- 29.Santa-Marta, M., F. A. da Silva, A. M. Fonseca, and J. Goncalves. 2005. HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J. Biol. Chem. 280:8765-8775. [DOI] [PubMed] [Google Scholar]
- 30.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 31.Simon, V., V. Zennou, D. Murray, Y. Huang, D. D. Ho, and P. D. Bieniasz. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathogens 1:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teutsch, S. M., D. R. Booth, B. H. Bennetts, R. N. Heard, and G. J. Stewart. 2003. Identification of 11 novel and common single nucleotide polymorphisms in the interleukin-7 receptor-alpha gene and their associations with multiple sclerosis. Eur. J. Hum. Genet. 11:509-515. [DOI] [PubMed] [Google Scholar]
- 33.Tian, C., X. Yu, W. Zhang, T. Wang, R. Xu, and X. F. Yu. 2006. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J. Virol. 80:3112-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, H., A. Sakurai, B. Khamsri, T. Uchiyama, H. Gu, A. Adachi, and M. Fujita. 2005. Unique characteristics of HIV-1 Vif expression. Microbes Infect. 7:385-390. [DOI] [PubMed] [Google Scholar]
- 35.Wichroski, M. J., K. Ichiyama, and T. M. Rana. 2005. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J. Biol. Chem. 280:8387-8396. [DOI] [PubMed] [Google Scholar]
- 36.Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wurtzer, S., A. Goubard, F. Mammano, S. Saragosti, D. Lecossier, A. J. Hance, and F. Clavel. 2006. Functional central polypurine tract provides downstream protection of the human immunodeficiency virus type 1 genome from editing by APOBEC3G and APOBEC3B. J. Virol. 80:3679-3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao, Z., E. Ehrlich, Y. Yu, K. Luo, T. Wang, C. Tian, and X. F. Yu. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349:290-299. [DOI] [PubMed] [Google Scholar]
- 39.Yang, B., L. Gao, L. Li, Z. Lu, X. Fan, C. A. Patel, R. J. Pomerantz, G. C. DuBois, and H. Zhang. 2003. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J. Biol. Chem. 278:6596-6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang, X., J. Goncalves, and D. Gabuzda. 1996. Phosphorylation of Vif and its role in HIV-1 replication. J. Biol. Chem. 271:10121-10129. [DOI] [PubMed] [Google Scholar]
- 41.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435-442. [DOI] [PubMed] [Google Scholar]
- 42.Yu, X., Y. Yu, B. Liu, K. Luo, W. Kong, P. Mao, and X. F. Yu. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056-1060. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L. Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.