

Abstract
In the present study we used a bioassay to study the effects of peroxynitrite (ONOO−) on angiotensin II (A-II)-triggered tension in isolated bovine coronary arteries in order to show the consequences of the previously reported PGI2-synthase inhibition by ONOO− in this model. The following results were obtained:
1 μmol L−1 ONOO− impaired A-II-induced vasorelaxation and caused a second long lasting constriction phase. Indomethacin (10−5M) prevented both effects. U51605, a dual blocker of PGI2-synthase and thromboxane (TX)A2-synthase mimicked the effects of ONOO−.
The selective TXA2/prostaglandin endoperoxide (PGH2) receptor antagonist SQ29548 antagonized the second vasoconstriction phase after ONOO−-treatment. Since a generation of TXA2 and 8-iso-prostaglandin F2α could be excluded a direct action of unmetabolized PGH2 on the TXA2/PGH2 receptor was postulated.
ONOO− dose-dependently inhibited the conversion of 14C-PGH2 into 6-keto-PGF1α in isolated bovine coronary arteries with an IC50-value of 100 nM.
Immunoprecipitation of 3-nitrotyrosine-containing proteins with a monoclonal antibody revealed PGI2-synthase as the only nitrated protein in bovine coronary arteries treated with 1 μmol l−1 ONOO−.
Using immunohistochemistry a co-localization of PGI2-synthase and nitrotyrosine-containing proteins was clearly visible in both endothelial and vascular smooth muscle cells. We concluded that ONOO− not only eliminated the vasodilatory, growth-inhibiting, antithrombotic and antiadhesive effects of PGI2 but also allowed and promoted an action of the potent vasoconstrictor, prothrombotic agent, growth promoter, and leukocyte adherer, PGH2.
Keywords: Peroxynitrite, tyrosine nitration, prostacyclin, prostaglandin, endoperoxide, superoxide anions, nitric oxide
Introduction
Vascular tone results from the finely tuned regulation of constriction and relaxation in vascular smooth muscle for which the endothelium acts as a sensor and signal transducer. Under physiological conditions, the endothelium plays a critical role in regulating the local blood supply by releasing the vasorelaxing factors nitric oxide (NO), prostacyclin (PGI2) and the not yet identified endothelium-derived hyperpolarizing factor (EDHF) as well as a series of vasoconstricting factors such as endothelins, prostaglandin H2 (PGH2), thromboxane A2 (TXA2) and leukotriene C4. These factors not only influence vascular tone but also affect haemostasis, thrombosis, vascular remodelling and the expression of chemotactic and adhesion molecules (Vane et al., 1990; Lüscher & Noll, 1995).
Superoxide anions (O2−) can exert a similar but indirect effect by trapping NO. O2− is generated through one-electron reduction of O2 by NAD(P)H oxidases, the respiratory chain in mitochondria and other enzymes such as xanthine oxidase (Laursen et al., 1997; Rajagopalan et al., 1996; White et al., 1996). Cultured endothelial cells produce O2− under basal conditions (Rosen & Freeman, 1984) and when stimulated such as through ischaemia/reperfusion (Zweier et al., 1987) or treatment with angiotensin-II (A-II) (Pueyo et al., 1998), bradykinin (Holland et al., 1990), calcium ionophore A23187 (Matsubara & Ziff, 1986a) or with cytokines such as interleukin−1 and interferon-γ (Matsubara & Ziff, 1986b). O2− per se is chemically stable and relatively unreactive. However, when it combines with NO at a nearly diffusion-controlled rate (Huie & Padjama, 1993) it forms peroxynitrite (ONOO−) (Beckman et al., 1990) as a potent oxidant capable of attacking DNA, lipids and proteins. ONOO− also can act as a powerful nitrating agent which nitrates tyrosine residues at the ortho position thereby irreversibly modifying proteins (Pryor & Squadrito, 1995). Since ONOO− is unstable and decomposes to nitrate and nitrite with a half-life less than 1 s in physiological solutions, nitration of tyrosine residues in proteins can be taken as footprints of ONOO− (Pryor & Squadrito, 1995).
Vascular cells are potential producers of ONOO− due to their capacity to simultaneously release O2− and NO. This is especially important under conditions when the production of these two radicals is upregulated. Indeed, nitrotyrosine has been detected in both endothelium and smooth muscle by nitrotyrosine-specific antibodies under conditions such as atherosclerosis (Beckman et al., 1994), acute lung injury in patients with septic shock (Kooy et al., 1995), rejected renal allografts (MacMillan-Crow et al., 1996), ischaemia-reperfusion in rat hearts (Wang & Zweier, 1996), as well as in lungs during acute endotoxaemia (Wizemann et al., 1994). It was also observed in brain vessels from CO-poisoned rats (Ischiropoulos et al., 1996) and endotoxin-treated rat aortae (Szabo et al., 1995). In perfused rat hearts, continued exposure of coronary arteries to ONOO− initially causes vasodilation which is progressively impaired (Villa et al., 1994). In addition, ONOO− promotes endothelium-dependent vasoconstriction after coronary reperfusion of the ischaemic heart (Pearson et al., 1991).
Recently, we reported that PGI2-synthase is inactivated by ONOO− (Zou & Ullrich, 1996) and that this irreversible inhibition is caused by nitration of an essential tyrosine at the active site (Zou et al., 1997). Moreover, inhibition by ONOO− was also observed in whole endothelial cells at submicromolar concentrations suggesting a possible role in vivo. In the present study, a bioassay was used to demonstrate that ONOO− selectively impaired PGI2-dependent vasorelaxation in bovine coronary arteries by preventing A-II-mediated PGI2 release via tyrosine nitration of PGI2-synthase. As a result, the remaining PGH2 further enhanced vasotonus by acting at the TXA2/PGH2 receptor. Our studies suggest that PGI2-synthase is the main target for ONOO− mediated nitration in bovine coronary arteries.
Methods
Isolation of bovine coronary arteries
Bovine hearts were obtained from the local slaughterhouse. The epicardial coronary arteries of the left ventricle were quickly dissected, cleaned of adhering fat and connective tissues and placed in an ice-cold Krebs-bicarbonate solution consisting of (in mM): NaCl 118; KCl 4.7; CaCl2 2.5; MgSO4 1.2; KH2PO4 1.2; NaHCO3 25; EDTA 0.016; glucose 11.1. The tissues were kept in ice-cold Krebs buffer for up to 24 h with frequent buffer changes.
Isometric measurement of tension in bovine coronary arteries
A spiral (20×5 mm) cut from bovine coronary arteries was suspended in a tissue bath filled with 15 ml of Krebs buffer, gassed with 95% O2, 5% CO2 (37°C; pH 7.4) and attached to a force-displacement TFT6V5 transducer coupled to a polygraph for the measurement of isometric tension. Passive tension was adjusted to approximately 1 g over a 30 min equilibrating period and then coronary arteries were constricted by addition of the thromboxane mimetic U46619 (0.001–0.01 μM). The vasorelaxation after acetylcholine (1 μM) was used to demonstrate the presence of endothelium. Throughout the experiment, care was taken to avoid any injury to the endothelium.
After rinsing the tissue with fresh buffer and allowing the mechanical tension to obtain a steady-state value, a reference response of vasocontraction-relaxation was obtained for each spiral by addition of A-II (50 nM). Thirty minutes after A-II stimulation, the medium was collected for prostanoid analysis and the tissue was treated with ONOO− as follows: the vessel was placed into a 2 ml Eppendorf tube containing 0.9 ml prewarmed Krebs buffer and 0.1 ml ONOO−, to give the indicated concentration of ONOO−, was quickly and thoroughly mixed. Control vessels were treated with the same volume of alkaline Krebs buffer medium or decomposed ONOO− (24 h at room temperature). Two minutes after treatment, the tissue was resuspended in the organ bath without adjusting the tension. Where required, the pharmacological agents indomethacin, SQ29548, CGS 13080, U51605 were added to the organ bath immediately after treatment. After 60 min of re-equilibration, the tissue was stimulated with the same concentration of A-II for another 30 min. At the end of the experiment, vessels were collected for immunoprecipitation and incubation media was collected and stored at −20°C for PG and TXA2 analysis. The percentage of vasoconstriction/relaxation was calculated as the percentage of relaxation by A-II in the secondary stimulation with A-II relative to that in the first stimulation. Similarly, the amounts of prostanoids were expressed as the percentage of prostanoids formed by the second stimulation compared to those after the first stimulation with A-II.
In order to evaluate the effects of ONOO− on the receptor-dependent or receptor-independent vasoconstriction mediated by U46619, PGH2 or KCl, a reference contractile response for each compound was initially obtained for each spiral. Subsequently, the tissues were treated with ONOO− or alkaline Krebs solution as described above and then resuspended in the organ bath containing fresh Krebs buffer. Once the tension had returned to baseline, the vessels were treated by addition of the same concentration of U46619 (5×10−8M), PGH2 (10−6M) or KCl (6×10−4M). Agonist-induced vasconstriction of coronary arteries was calculated as the percentage of vasoconstriction in the secondary stimulation compared to the reference contractile response.
Extraction and analysis of prostanoids
After acidification with 1 N HCl to pH 3.5 the collected medium was extracted with three volumes of ethyl acetate. The organic phases were collected and evaporated to dryness under nitrogen. Samples were resuspended in phosphate-buffered-saline (PBS) containing 0.1% bovine serum albumin (BSA). Prostanoids and thromboxane B2 were analysed by enzyme-linked immunoassay kits, according to the instructions provided by the supplier.
Activity assay of PGI2-synthase
Bovine coronary spirals (20×5 mm) stored in Krebs buffer were treated with the indicated concentrations of ONOO− and were quickly and thoroughly mixed. Two minutes after treatment, 100 μM [14C]-PGH2 was added and the samples were incubated for additional 3 min. The reaction was stopped by acidification with 1 N HCl to pH 3.5. The incubation media were extracted with three volumes of ethyl acetate and after centrifugation, the organic phases were evaporated to dryness under nitrogen. Samples were then resuspended in 60 μl of ethyl acetate and subsequently separated by TLC (ethyl acetate/water/isooctane/acetic acid; 90:100:50:20). Prostanoids were quantified with a phosphor imager system (Image Quant, Molecular Dynamics) as previously described (Zou & Ullrich, 1996).
Immunoprecipation of nitrotyrosine-containing proteins
The vessels collected from the organ bath were homogenized in a buffer consisting of (in mM) Tris 50, EDTA 1, BHT 0.1, PMSF (pH 7.4) 1 and centrifuged (12,000×g) for 1 h. Supernatants were diluted in Tris buffer (50 mM) containing (in mM) PMSF 1, EDTA 5, NaCl 150, and 0.5% Nonidet-40 (pH 8.0). Solubilized extracts were sonicated for 30 s in a Branson Sonifier 250 (Schwäbisch Gmünd, Germany), setting 5, at 50% duty cycle and centrifuged (14,000×g) at 4°C for 5 min to remove cellular debris. Protein concentrations were determined using the Bradford assay (Bradford, 1976).
Solubilized proteins (3 mg) were precleared by addition of 40 μl of protein A sepharose CL-4B and the supernatant was incubated (18 h, 4°C) with 10 μg of monoclonal anti-nitrotyrosine antibody (Upstate Biotechnology). Immune complexes were precipitated with 30 μl of protein A sepharose CL-4B and washed with 0.5 ml SNNTE (0.5% sucrose, 1% NP-40, NaCl (0.5 mM), Tris (50 mM), EDTA, pH 7.4 (5 mM). Resuspended protein pellets were then precipitated by centrifugation (14,000×g, 1 min) and resuspended in 40 μl Laemmli sample buffer/β-mercaptoethanol (9 : 1), heated at 95°C for 5 min and kept on ice.
Western blots
Protein precipitates were separated by 7.5% SDS–PAGE (30 mA, 1 h) and blotted for 1 h with a constant current of 200 mA onto a nitrocellulose membrane in a semidry blot procedure (48 mM Tris/39 mM glycine/20% methanol/0.037% SDS). Proteins were visualized with a 0.1% Ponceau S solution in 5% acetic acid to check transfer efficiency. After destaining, the membrane was blocked with 5% milk powder in PBS/ 0.1% Tween 20 for 2 h at room temperature and incubated with a polyclonal antibody directed against PGI2-synthase (Siegle et al., 1994) (1 μg ml−1) overnight at 4°C. After washing several times with PBS/0.1% Tween 20, the membrane was further incubated with a HRP-conjugated goat anti-rabbit antibody at a dilution of 1 : 7500 for 45 min. Antibody binding was visualized by the ECL technique, according to the instructions of the supplier (Amersham). The blots which had been stained with a polyclonal antibody against PGI2-synthase were then stripped by incubating the membrane in stripping buffer (100 mM 2-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7) at 50°C for 30 min with agitation. The membranes were washed, blocked, and incubated overnight at 4°C with a monoclonal antibody against 3-nitrotyrosine at a dilution of 1 μg ml−1 and with a HRP-conjugated goat anti-mouse antibody for 45 min at a dilution of 1 : 7500. Antibody binding was visualized by the ECL technique.
Immunohistochemistry
Bovine coronary arteries were treated with various concentrations of ONOO− or the same volume of alkaline Krebs buffer for control tissues, as described above. Arteries were then fixed in 4% paraformaldehyde for 3 h followed by cryoprotection at 1 h incubations in 10, 20, and 30% sucrose in 0.1 M sodium cacodylate buffer. Tissues were then embedded in pre-cooled, OCT-containing cups and frozen in liquid nitrogen-cooled isopentane. Samples were stored at −80°C. Nine μm sections were mounted on poly-L-lysine coated slides, air-dried and blocked for 45 min in a blocking solution consisting of 4% fatty acid-free BSA, either 10% goat serum or 10% horse serum and 3% Triton X-100 in PBS, pH 7.2. Sections intended for double staining and the appropriate controls were coincidently blocked with both serum type blocking solutions. This was followed by coincubation with the monoclonal anti-nitrotyrosine antibody (15 μg ml−1) and the polyclonal PGI2-synthase antibody (5 μg ml−1) overnight at 4°C. Control sections were stained with the nitrotyrosine antibody prepared in 10 mM 3-nitrotyrosine in 0.1 M PBS. Antibody binding was then visualized by coincubation of fluorescein isothiocyanate-conjugated anti-mouse IgG (1 : 50 dilution) with anti-rabbit IgG conjugated to Texas Red (1 : 100 dilution) for 1 h. Sections were washed and examined under a Leica (Leica, Wetzlar, Germany) microscope. Negative controls were performed by either eliminating one or the other or both primary antibodies, or by incubating sections with a non-specific primary antibody. All pictures were obtained under 400 fold magnification with identical camera and print settings.
Reagents
[14C]-AA (53 mCi mmol−1) was purchased from Dupont (Dreieich, Germany). 11-dideoxy-9α, 11α-methanoepoxy prostaglandin F2α (U46619), carbethoxyhexyl imidazol, 5-heptenoic acid,7[6-(1-octenyl)-2,3-diazabicyclo[2.2.1]hept-2-en-5-yl-,[1R-[1α, 4α, 5β (Z), 6α (E) ] ]-(U51605), 1S[1α, 2B (5Z), 3b, 4α]-7-[3- [2 (phenylamino) carbonyl] hydrazino methyl[-7-oxabicyclo(2.2.1)hept-2-yl-5-heptenoic acid (SQ29548), indomethacin, Nω-mono-methyl-L-arginine (L-NMMA), PGI2, diethylamide NONOate (DEA-NO), and the enzyme-linked immuno-assay kits for 6-keto-PGF1α, PGE2, TxB2, PGF2α, and 8-iso-PGF2α were obtained from Cayman SPI (Massy, France). Losartan was acquired from Merck (Darmstadt, Germany). Imidazol [1,5α] pyridine-5-hexanoic acid (CGS13080) was kindly donated by Ciba-Geigy (Basel, Switzerland). Protein-A-sepharose CL-4B was obtained from Pharmacia (Freiburg, Germany). A monoclonal antibody against 3-nitrotyrosine was obtained from Upstate Biotechnology Incorporated (Hamburg, Germany). A-II, bovine serum albumin (BSA), sodium cacodylate, 3-nitrotyrosine, 3-amino-L-tyrosine, O-phospho-DL-tyrosine and 3-chloro-L-tyrosine were purchased from Sigma (Deisenhofen, Germany). OCT compound embedding medium was purchased from Miles (Elkhart, IN, U.S.A.). Goat serum, horse serum, fluorescein-labelled anti-mouse IgG (H+L) (horse anti-mouse, rat adsorbed) were acquired from Camon (Wiesbaden, Germany). Texas Red-conjugated Affinipure F(ab′)2 fragment was obtained from Jackson Immunoresearch, Dianova (Hamburg, Germany). A rat anti-rabbit IgG (H+L), a goat anti-rabbit IgG, a horseradish peroxidase conjugated IgG and a HRP-conjugated goat anti-mouse IgG were purchased from Pierce (Freiburg, Germany). The ECL kits and nitrocellulose membranes (Hybond-C) were purchased from Amersham (Braunschweig, Germany). Organic solvents were obtained from Merck (Darmstadt, Germany). Other chemicals not indicated, were acquired from the local supplier.
ONOO− and [14C]-PGH2 were prepared as described previously (Zou & Ullrich, 1996; Zou et al., 1997). A ONOO− stock solution was diluted in alkaline Krebs-bicarbonate buffer (pH=14) on ice immediately prior to use in order to avoid tissue pH and osmolar shifts during ONOO− treatment.
Results
Mediators of angiotensin II dependent coronary artery vasoconstriction and relaxation
Exogenous addition of PGI2 (10−8–10−6μM) and NO from the NO-releasing agent, DEA-NO (10−8–10−6μM) caused a dose-dependent vasorelaxation in the vessels which had been preconstricted by the PGH2 analogue U46619 (5×10−8μM) (data not shown). Exogenous O2− generated from xanthine oxidase (0.1–100 mU mL−1) and hypoxanthine (10−4M) did not affect the basal vasotone or U46619 induced vasoconstriction. As seen in Figure 1, A-II evoked a strong vasoconstriction rapidly followed by vasorelaxation of the bovine coronary arteries. Vasoconstriction was selectively blocked by Losartan (5×10−7M), a selective AT-1 receptor blocker (not shown). The endothelial NO-synthase inhibitor L-NMMA (10−4M) evoked a slight decrease in vasorelaxation (−25.3±2.1%). Indomethacin (10−5M) did not inhibit A-II mediated vasoconstriction suggesting that cyclo-oxygenase activity was not necessary for the initial A-II-induced vasoconstriction. However, in the presence of indomethacin, A-II-induced vasorelaxation was selectively suppressed (−74.6±6.5%) (Figure 1).
Figure 1.

Representative recordings of A-II-triggered vasoconstriction and relaxation in bovine coronary arteries: effects of indomethacin, U51605, and L-NMMA. Data are expressed as means±s.e.mean from 12 separate experiments. Δ indicates the addition of 5×10−8M A-II. ▿ indicates the addition of 10−5M SQ29 548. (a) Control tissue; (b) 10−4M L-NMMA; (c) 10−5M indomethacin; (d) 5×10−6M U51605.
This was paralleled by a decrease in 6-keto-PGF1α release (−73.6±6.5%) thereby confirming the predominant role of PGI2 for the relaxation phase. After A-II stimulation, large quantities of prostaglandins were detected in the supernatants, the main product being 6-keto-PGF1α.
Effect of PGI2-synthase inhibitor U51605 on A-II- induced vasoconstriction/relaxation and prostanoid release
U51605 (10−7–10−4M), a selective inhibitor of PGI2-synthase as well as of TXA2 synthase (Gorman et al., 1979), did not modify the A-II-induced vasoconstriction. Rather, as shown in Figure 1, it selectively impaired the relaxation phase and triggered a second phase of vasoconstriction with an IC50-value of about 2×10−6M. Indomethacin (10−5M) effectively restored the U51605 impaired vasorelaxation and abolished the second vasoconstriction (data not shown). SQ29548(10−5M), a selective antagonist of the TXA2/PGH2 receptor (Ogletree et al., 1985) which neither had an effect on A-II-evoked vasoconstriction nor the basal vasotone, competitively antagonized the U51605-specific rise in tension (−76.3±4.4%). In contrast, the selective TXA2-synthase inhibitors CGS 13080(10−5M) (McNab et al., 1984) and carbethoxyhexyl imidazol (10−5M), had no effect on the second phase of vasoconstriction (data not shown).
Analysis of A-II released prostanoids in the supernatant confirmed the potent inhibition of PGI2 and TXA2 synthesis by 5×10−6M U51605 (PGI2: 1854±214 vs 498±87 pg ml−1= 73.1%; TXA2: 149±21 vs 37±15 pg ml−1= 75.2%) (Table 1).
Table 1.
Effects of U51605 and peroxynitrite on angiotensin II-stimulated release of prostanoids in bovine coronary arteries

Interestingly, the level of PGE2, a presumed hydrolysis product of PGH2, was largely increased after U51605 treatment (496±77 vs 2008±137 pg ml−1=304.2%). This pharmacological behaviour suggests that the second constriction phase could be due to the accumulation of PGH2 as a result of PGI2-synthase inhibition.
Effect of peroxynitrite on A-II induced vasoconstriction/vasorelaxation and prostanoid release
In this well characterized pharmacological model, the effects of ONOO− on A-II-triggered and PGI2-dominant vasorelaxation were examined (Figure 2).
Figure 2.

Effects of peroxynitrite on A-II-mediated vasoconstriction/relaxation in bovine coronary arteries: TXA2/PGH2 receptor-mediated vasospasm in ONOO− treated bovine coronary arteries identified by indomethacin and SQ29548. Results are expressed as means±s.e.mean from ten separate experiments. Δ indicates the addition of 5×10−8M A-II. ▿ indicates the addition of 10−5M SQ29 548. (a) Tissue treated with decomposed ONOO−; (b) ONOO− (10−6M); (c) ONOO− (10−6M) in the presence of SQ 29548 (10−5M); (d) ONOO− (10−6M) in the presence of indomethacin (10−5M); (e) ONOO− (10−6M) in the presence of CGS13080 (10−5M).
As shown in Figure 2, 10−6M ONOO− neither altered the resting vascular tone nor the initial A-II mediated vasoconstriction. However, the same concentration of ONOO− selectively suppressed the A-II-mediated vasorelaxation phase (−81± 11%). Shortly thereafter, a secondary rise in vasotension was apparent. As was seen in the presence of U51605, the addition of indomethacin also abolished the second phase of vasoconstriction, in agreement with the participation of cyclo-oxygenase-derived prostanoids. Involvement of the TXA2/PGH2 receptor in the delayed vasoconstriction was indicated by addition of SQ29548, which competitively blunted this phase. Since both CGS13080 (10−5M) and carbethoxyhexyl imidazol (10−5M) were ineffective in suppressing vasoconstriction, enhanced synthesis of TXA2 could be excluded. Taken together, it is reasonable to postulate that like U51605, ONOO− could have blocked PGI2-synthase and accumulating, unmetabolized PGH2 may have reacted with the TXA2/PGH2 receptor. Suppressed levels of 6-keto-PGF1α in the incubation media (2035±225 vs 206±33 pg ml−1=−89%, P<0.001) further support this hypothesis. Indeed, levels of PGE2, a presumed metabolite of PGH2, were dramatically increased (396±77 vs 2237±245 pg ml−1=464%, P<0.001) (Table 1). Decomposed ONOO− altered neither the A-II-mediated vasorelaxation nor the release of 6-keto-PGF1α and PGE2. The amounts of PGF2α and TxB2 remained unchanged (PGF2α: 276±42 vs 310±56 pg ml−1; TxB2: 189±41 vs 167± 35 pg ml−1) (Table 1) after ONOO− treatment thereby supporting the hypothesis that unmetabolized PGH2 after PGI2-synthase inhibition was converted to PGE2. PGH2 could have activated its own receptor before being metabolized to PGE2.
The level of PGH2 in vascular tissues is determined by the balance between the rate of PGH2 production through cyclo-oxygenase and the conversion of PGH2 into PGI2, PGE2 or other prostanoids. Since the total cyclo-oxygenase activity remained unchanged (±1%, see Table 1), excess formation of PGH2 by cyclo-oxygenase could be excluded. 8-iso-PGF2α production was not affected by ONOO− treatment (32±6 vs 35±7, P>0.5) thereby ruling out lipid peroxidation after this low-dose ONOO− treatment (Table 1).
Effects of peroxynitrite on U46619- and KCl- induced vasoconstriction
Enforced vasoconstriction could have been caused by an increased sensitivity of vascular smooth muscle to vasoconstricting factors. U46619, a synthetic PGH2 analogue which acts as a TXA2/PGH2 receptor agonist, elicited a similar constrictor response in both ONOO−-treated (1–100 μM) and in untreated tissues (Figure 2). Therefore, the possibility of an increased response of the TXA2/PGH2 receptor under the influence of ONOO− could be excluded. 60 mM KCl elicited identical responses of isometric tension in both ONOO−-treated and untreated vessels (data not shown).
Effect of peroxynitrite on the conversion of [14C]-PGH2 to PGI2 and PGE2 in bovine coronary arteries
To directly verify the inhibition of PGI2-synthase by ONOO−, coronary arteries pretreated with varying concentrations of ONOO− were incubated with [14C]-PGH2 and the production of radioactive 6-keto-PGF1α was monitored. A dose-dependent inhibition of 6-keto-PGF1a production was seen in vessels treated with ONOO− (10−8–10−5M) and occurred concomitantly with an increase in PGE2. These results further confirmed the presence of considerable amounts of PGI2-synthase in bovine coronary arteries and that ONOO− selectively suppressed the conversion of PGH2 into PGI2 (Figure 3).
Figure 3.

Effect of peroxynitrite on [14C]-PGH2-dependent prostanoid production in bovine coronary arteries. 220–250 mg of vessel tissue were treated with various concentrations of ONOO− or the same volume of decomposed ONOO−. Five minutes after treatment, 100 μM [14C]-PGH2 was added and incubated for 3 min. The reaction was stopped by acidification to pH 3.5. Prostanoids were extracted and analysed as described under Methods. Results are means±s.e.mean from five separate experiments.
Identification of nitrotyrosine in PGI2 synthase
In our previous studies with isolated PGI2-synthase, we presented evidence that the inhibition of enzyme activity by ONOO− occurred in parallel with positive staining for antibodies against both PGI2-synthase and 3-nitrotyrosine in Western blot analysis (Zou et al., 1997). In the present study, 3-nitrotyrosine-containing proteins from whole vessels were precipitated with a monoclonal antibody against 3-nitrotyrosine. As also shown in Figure 4, such precipitates reacted positively with a polyclonal antibody against PGI2-synthase.
Figure 4.
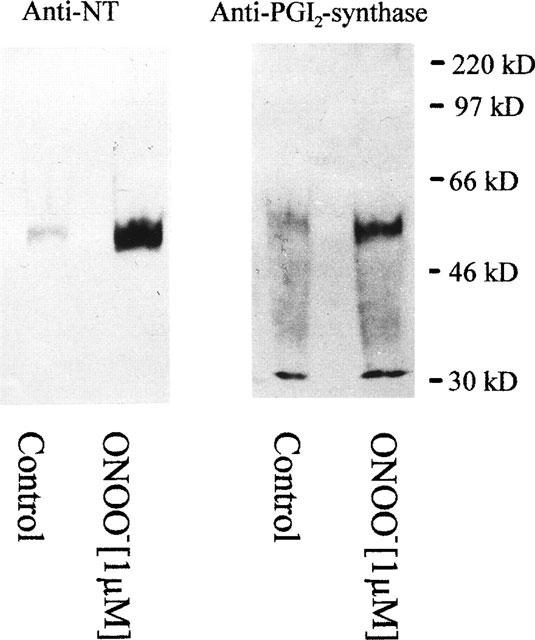
Western blot analysis of bovine coronary arteries treated with peroxynitrite (1 μM). Blots were stained with the monoclonal antibody against 3-nitrotyrosine (NT) and the polyclonal antibody against PGI2-synthase. After treatment, bovine coronary arteries were homogenized. 3-nitrotyrosine-containing proteins were precipitated by a monoclonal antibody against 3-nitrotyrosine, separated by SDS–PAGE, blotted onto a nitrocellulose membrane and detected as described under Methods. Results are representative for experiments with bovine coronary arteries from 8–10 hearts.
In these Western blots a main band appeared at 52 kD which co-stained with antibodies against PGI2-synthase as well as against nitrotyrosine. A second band at 30 kD was detected by a polyclonal antibody against PGI2-synthase but although it was precipitated by the nitrotyrosine antibody it did not stain with the same antibody in the plot of Figure 4. However, this may be due to lower affinities of monoclonal antibodies compared with polyclonal antiserum used for the detection of PGI2-synthase. In fact, under conditions of higher loading of the gels or longer exposure times the 30 kD band emerged from a high background. A possible staining of IgG light chains at this position was excluded by the appropriate controls with the secondary antibody (goat-anti-rabbit-HRP-conjugate) alone.
Immunohistochemistry of tyrosine-nitrated proteins and of PGI2-synthase
Dense staining by anti-PGI2-synthase antibody was visible in the endothelium. In addition, a clear wave-like pattern was seen in vascular smooth muscle cells (Figure 5a and d).
Figure 5.

Co-localization of 3-nitrotyrosine positive proteins and PGI2-synthase in bovine coronary arteries treated with 1 μM or decomposed peroxynitrite. The yellow colourings resulting from a computer-generated overlay of green and red fluorescence indicate areas of the co-localization of anti-PGI2 synthase and anti-nitrotyrosine staining in bovine coronary arteries with or without ONOO− treatment (400 fold magnification). (a) PGI2-synthase stainings in untreated bovine coronary arteries. (b) Computer-generated overlay of red fluorescence (PGI2-synthase staining; a) and green (anti-nitrotyrosine staining; c) resulting from double staining in untreated bovine coronary arteries (overlay of a and c). (c) Anti-nitrotyrosine staining in untreated vessels. (d) PGI2-synthase staining in 1 μM ONOO−-treated vessels. (e) The computer-generated overlay of red (anti-PGI2-synthase staining; d) and green fluorescence (anti-nitrotyrosine staining; f) resulting from double staining in 1 μM ONOO− treated vessels (overlay of d and f). (f) Anti-nitrotyrosine staining in 1 μM ONOO−-treated vessels. (g) Texas red and FITC alone (anti PGI2-synthase and anti-nitrotyrosine omitted). (h) Anti-nitrotyrosine staining in ONOO− treated vessels but in the presence of 10 μM nitrotyrosine.
The obvious difference of staining by anti-PGI2-synthase antibody in the vessels after ONOO− treatment could be due to an increased degradation of PGI2-synthase after nitration (unpublished data). No staining was observed when the anti-PGI2-synthase antibody was omitted (data not shown). In untreated tissues, α-nitrotyrosine staining was only weakly visible (Figure 5c) yet became especially apparent in areas of focal intimal thickening (data not shown). ONOO−(1 μM) treatment clearly increased 3-nitrotyrosine staining in both endothelium and vascular smooth muscle cells as compared to untreated vessels (Figure 5f). FITC-staining under all conditions resulted in heavy staining of the basal membrane. The specificity of the antibody against 3-nitrotyrosine was supported by incubation of the anti-nitrotyrosine antibody in 10 mM nitrotyrosine when anti-nitrotyrosine staining was blocked in ONOO− treated tissues (Figure 5h) but anti-PGI2-synthase staining remained clearly visible (data not shown). Neither 3-amino-L-tyrosine (10 mM), 3-chloro-L-tyrosine (10 mM) nor O-phospho-DL-tyrosine (10 mM) blocked 3-nitrotyrosine staining in ONOO−-treated tissues (data not shown).
Co-localization of anti-PGI2-synthase and anti-nitrotyrosine in the endothelium are clearly visible in Figure 5d and f, respectively, where identical staining patterns with the two antibodies are evident. Moreover, a computer-generated overlay of green and red fluorescence provided a yellow colouring to indicate areas of co-localization (Figure 5e). In the smooth muscle layer the overlap is not complete but cells stained in green are present also in the controls.
Discussion
The use of aortic rings in a physiological bath solution coupled to an isometric force measurement device is a well-established model for pharmacological and physiological studies on vascular tone. In bovine coronary arteries, A-II acts at the AT-1 receptor causing vasoconstriction. This is counterregulated by the release of a main vasodilator identified as PGI2. Analogous to the action of the TXA2 and PGI2-synthase inhibitor U51605, 1 μM ONOO− not only abolished this PGI2-mediated vasorelaxation but resulted in a second phase of vasoconstriction. This second phase was inhibited by a cyclo-oxygenase inhibitor as well as in the presence of the selective TXA2/PGH2 receptor antagonist SQ29548. Since TXA2 was ruled out as a possible mediator, it is reasonable to postulate that non-metabolized PGH2, which accumulated following PGI2 inhibition, caused the second phase of vasoconstriction. In support of this, 6-keto-PGF1α levels were suppressed in parallel with the increased vasoconstrictory action whereas levels of PGE2, an enzymatic or non-enzymatic metabolite of PGH2, were increased.
The presence of PGI2-synthase in bovine coronary arteries was further confirmed by the conversion of [14C]-PGH2 to labelled 6-keto-PGF1α. In isolated bovine coronary arteries, the selective inhibition of PGI2-synthase by ONOO− was seen by a decreased transformation of [14C]-PGH2 into 6-keto-PGF1α and by a shift to PGE2 (Figure 3). The above results therefore support a reaction of PGH2 at the smooth muscle TXA2/PGH2 receptor followed by conversion into PGE2.
Alternatively the secondary phase of increased vasoconstriction might also have been related to an increased sensitivity of vascular smooth muscle to a vasoconstrictory prostanoid. However, as shown by the experiment with U46619, up to 100 μM of ONOO− failed to increase PGH2 receptor sensitivity. The constriction responses to A-II and KCl were not affected by ONOO− treatment, suggesting that ONOO−-induced vasospasm was not due to a nonspecific effect of ONOO− on receptor-mediated or receptor-independent vasoconstriction.
Another metabolite which may have been responsible for the increased vasoconstriction is 8-iso-PGF2α, a non-cyclo-oxygenase-derived prostanoid. 8-iso-PGF2α triggers vascular constriction either by a separate receptor (Takahashi et al., 1992) or by the TXA2/PGH2 receptor (Laskey & Mathew, 1996) and ONOO− has been reported to increase 8-iso-PGF2α production through lipid peroxidation (Crow et al., 1997). However, the amount of 8-iso-PGF2α produced in bovine coronary arteries was minimal and its production was not influenced by the employed concentrations of ONOO−. Therefore, it is also unlikely that 8-iso-PGF2α was responsible for the increased vasoconstriction seen in vessels treated with ONOO−.
ONOO− has been characterized as an oxidizing and nitrating agent (Huie & Padjama, 1993; Beckman et al., 1990; 1994; Pryor & Squadrito, 1995; Kooy et al., 1995; MacMillan-Crow et al., 1996; Wang & Zweier, 1996; Wizemann et al., 1994; Ischiropoulos et al., 1996; Szabo et al., 1995). Having reported that ONOO− selectively inactivated PGI2-synthase by a mechanism of tyrosine nitration in bovine aortic microsomal fractions (Zou et al., 1997), we now extended our studies by using immunohistochemical and immunoprecipation techniques. A concentration of 1 μM ONOO− strongly increased nitrotyrosine staining in both endothelium and smooth muscle (Figure 5f). Co-localization of nitrotyrosine and PGI2-synthase was clearly visible (Figure 5e). Moreover, Western blots of immunoprecipitates stained with a monoclonal antibody against 3-nitrotyrosine showed one band at 52 kD which co-stained with a polyclonal antibody against PGI2-synthase (Figure 4). Polyclonal PGI2-synthase antibodies detected an additional band with a molecular weight of 30 kD. Since it was precipitated and could be detected by a monoclonal antibody against 3-nitrotyrosine (data not shown). It is reasonable to suggest that it might be a degradation product of nitrated PGI2-synthase. Purified PGI2-synthase after ONOO− treatment also results in this band together with a 46 kD band (unpublished data) indicating a stepwise degradation after nitration. Nitrated Mn-SOD has been reported in rejected kidney allografts (MacMillan-Crow et al., 1996) and in brain tissue (Lin et al., 1994) but was not detected in our present experiments.
In 3-nitrotyrosine immunoprecipitates PGI2-synthase was identified as the major protein. Interestingly, the untreated control tissue also contained nitrated PGI2-synthase in agreement with a weak staining also by immunohistochemistry. Since this staining was co-localized with PGI2-synthase it is justified to assume that a nitration had been occurred already in vivo or ex vivo before the incubations in the organ baths.
Impaired vasorelaxation and increased endothelium-derived cyclo-oxygenase-dependent vasoconstriction associated with decreased PGI2-synthesis have been widely described in different animal models of vascular diseases including hypertension (De Côrtes et al., 1996), atherosclerosis (Siegel et al., 1992) and diabetes (Tesfamariam, 1994). PGH2, PGH2-derived TXA2 and O2− have been referred to as the mediators of endothelium-dependent vasoconstriction (Katusic & Vanhoutte, 1989; Tesfamariam & Cohen, 1992; Katusic, 1996) and both the TXA2/PGH2 receptor antagonist and SOD were effective in restoring impaired endothelium-dependent relaxations (Tesfamariam & Cohen, 1992; Katusic, 1996; Lin et al., 1991; Rodrigo et al., 1997; Taddei et al., 1993) suggesting that O2− and arachidonic acid-derived contracting factors are important in endothelial dysfunction. Moreover, evidence exists that vasoconstricting prostanoids contribute to the impaired acetylcholine-derived vasorelaxation observed in the forearm circulation of patients with essential hypertension (Taddei et al., 1993), in human coronary arteries (Siegel et al., 1992; Zeiher et al., 1993; Ludmer et al., 1986) and in microvessels of patients with coronary atherosclerosis (Treasure et al., 1991) since a normal response is restored after treatment with a cyclo-oxygenase inhibitor (Taddei et al., 1993; Treasure et al., 1991). Although overproduction of oxygen radicals is a well known cause of increased vasoconstriction, O2− itself lacks a direct vasoconstrictor effect (Katusic & Vanhoutte, 1989; Tesfamariam & Cohen, 1992; Katusic, 1996; Lin et al., 1991). Our results offer an explanation of how O2−, via ONOO−, triggers cyclo-oxygenase-dependent vasoconstriction.
In summary, we present evidence that 1 μM of ONOO− inhibit PGI2-synthase in bovine coronary arteries and that the remaining PGH2 acts on the TXA2/PGH2 receptor before it is converted to PGE2. The inhibition of PGI2-synthase was accompanied by a tyrosine nitration in agreement with earlier in vitro results. The fact that nitrotyrosine staining was already detectable in minor amounts without ONOO− treatment could be taken as indication that the process of nitration had started in vivo or ex vivo by the simultaneous formation of O2− and NO in the endothelium. Recent results on IL-1 treated rat mesangial cells (Zou et al., 1998) also allow the conclusion that the required ONOO− levels for PGI2-synthase inhibition are generated under pathophysiological conditions like inflammation or ischaemia/reperfusion. It therefore should be considered that O2− acts as a pathophysiological mediator to suppress first NO and secondarily PGI2 actions.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft. We thank Dr Marcel Leist for kindly generating computer photographs and Mrs Naschwitz for preparing the manuscript.
Abbreviations
- A-II
angiotensin II
- BSA
bovine serum albumin
- ECL
enhanced chemiluminescence
- EDHF
endothelium derived hyperpolarizing factor
- L-NMMA
Nω-mono-methyl-L-arginine
- NO
nitric oxide
- O2−
superoxide anion
- ONOO−
peroxynitrite
- PBS
phosphate buffered saline
- PG
prostaglandin
- PGI2
prostacyclin
- SDS–PAGE
polyacrylamide gel electrophoresis in presence of sodium dodecyl sulphate
- SOD
superoxide dismutase
- TXA2
thromboxane A2
References
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.A., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKMAN J.S., YE Y.Z., ANDERSON P.G., CHEN J., ACCAVETTI M.A., TARPEY M.M., WHITE C.R. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe-Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Ann. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- CROW J.P., SAMPSON J.B., ZHUANG Y., THOMPSON J.A., BECKMAN J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- DE CÔRTES F.S., ANDRIANTSITOHAINA R., STOCLET J.C. Alternations of cyclo-oxygenase products and NO in response to angiotensin II of resistance arteries from the spontaneously hypertensive rat. Brit. J. Pharmacol. 1996;119:1635–1641. doi: 10.1111/j.1476-5381.1996.tb16083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GORMAN R.R. , HAMILTON R.D., HOPKINS N.K. Stimulation of human foreskin fibroblast adenosine 3′, 5′-cyclic monophosphate levels by prostacyclin (prostaglandin I2) J. Biol. Chem. 1979;254:1671–1676. [PubMed] [Google Scholar]
- HOLLAND J.A., PRITCHARD K.A., PAPPOLLA M.A., WOLIN M.S., ROGERS N.J., STEMERMAN M.B. Bradykinin induces superoxide anion release from human endothelial cells. J. Cell. Physiol. 1990;143:21–25. doi: 10.1002/jcp.1041430104. [DOI] [PubMed] [Google Scholar]
- HUIE R.E., PADJAMA S. The reaction of NO with superoxide. Free Radical Res. Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- ISCHIROPOULOS H., BEERS M., OHNISHI S.T., FISCHER D., GARNER S.E., THOM S.R. Nitric oxide production and perivascular tyrosine nitration in brain after carbon monoxide poisoning in the rat. J. Clin. Invest. 1996;97:2260–2267. doi: 10.1172/JCI118667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATUSIC Z.S. Superoxide anion and endothelial regulation of arterial tone. Free Radical Biol. Med. 1996;20:443–448. doi: 10.1016/0891-5849(96)02116-8. [DOI] [PubMed] [Google Scholar]
- KATUSIC Z.S., VANHOUTTE P.M. Superoxide anion is an endothelium-derived contracting factor. Am. J. Physiol. 1989;257:H33–H37. doi: 10.1152/ajpheart.1989.257.1.H33. [DOI] [PubMed] [Google Scholar]
- KOOY N.W., ROYALL J.A., YE Y.Z., KELLY D.R., BECKMAN J.S. Evidence for in vivo peroxynitrite production in human acute lung injury. Am. J. Respir. Crit. Care Med. 1995;151:1250–1254. doi: 10.1164/ajrccm/151.4.1250. [DOI] [PubMed] [Google Scholar]
- LASKEY R.E., MATHEW W.R. Nitric oxide inhibits peroxynitrite-induced production of hydroxyeicosatetraenoic acids and F2-isoprostanes in phosphatidylcholine liposomes. Arch. Biochem. Biophys. 1996;330:115–124. doi: 10.1006/abbi.1996.0242. [DOI] [PubMed] [Google Scholar]
- LAURSEN J.B., RAJAGOPALAN S., TARPEY M.M., FREEMAN B.A., HARRISON D.G. A Role of superoxide in angiotensin II but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- LIN L., BALAZY M., PAGANO P.J., NASJLETTI A. Expression of prostaglandin H2-mediated mechanism of vascular contraction in hypertensive rats: Relation to lipoxygenase and prostacyclin synthase activities. Circ. Res. 1994;74:197–205. doi: 10.1161/01.res.74.2.197. [DOI] [PubMed] [Google Scholar]
- LIN L., MISTRY M., STIER C.T., NASJLETTI A. Role of prostanoids in renin-dependent and renin-independent hypertension. Hypertension. 1991;17:517–525. doi: 10.1161/01.hyp.17.4.517. [DOI] [PubMed] [Google Scholar]
- LUDMER P.L., SEWYN A.P., SHOOK T.L., WAYNE R.R., MUDGE G. H., ALEXANDER R.W., GANZ P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- LÜSCHER T.F., NOLL G. The pathogenesis of cardiovascular disease: role of the endothelium as a target and mediator. Atherosclerosis. 1995;118:S81–S90. [PubMed] [Google Scholar]
- MACMILLAN-CROW L.A., CROW J.P., KERBY J.D., BECKMAN J.S., THOMPSON A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUBARA T., ZIFF M. Superoxide anion release by human endothelial cells: Synergism between a phorbol ester and a calcium ionophore. J. Cell Physiol. 1986a;127:207–210. doi: 10.1002/jcp.1041270203. [DOI] [PubMed] [Google Scholar]
- MATSUBARA T., ZIFF M. Increased superoxide anion release from human endothelial cells in response to cytokines. J. Immunol. 1986b;137:3295–3298. [PubMed] [Google Scholar]
- MCNAB M.W., FOLTZ E.L., GRAVES B.S., RINEHART R.K., TRIPP S.L., FELICIANO N.R., SEN S. The effects of a new thromboxane synthetase inhibitor CGS13080 in man. J. Clin. Pharmacol. 1984;24:76–83. doi: 10.1002/j.1552-4604.1984.tb02768.x. [DOI] [PubMed] [Google Scholar]
- OGLETREE M., HARRIS D.N., GREENBERG R., HASLANGER M.F., NAKANE M. Pharmacological actions of SQ29548, a novel selective thromboxane antagonist. J. Pharmacol. Exp. Ther. 1985;234:435–441. [PubMed] [Google Scholar]
- PEARSON P.J., LIN P.J., SCHAFF H.V. Production of endothelium-derived contracting factor is enhanced after coronary reperfusion. Ann. Thorac. Surg. 1991;51:788–793. doi: 10.1016/0003-4975(91)90126-b. [DOI] [PubMed] [Google Scholar]
- PRYOR W.A., SQUADRITO G.L. The chemistry of peroxynitrite: a product of the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- PUEYO M.E., ARNAL J.F., RAMI J., MICHEL J.B. Angiotensin-II stimulated the production of NO and peroxynitrite in endothelial cells. Am. J. Physiol. 1998;274:C214–C220. doi: 10.1152/ajpcell.1998.274.1.C214. [DOI] [PubMed] [Google Scholar]
- RAJAGOPALAN S., KURZ S., MUNZEL T., TARPEY M., FREEMAN B.A., GRIENDLING K.K., HARRISON D.G. Angiotensin-II mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGO E., MAESO R., MUNOZ-GARCIA R., NAVARRO-CID J., RUILOPE L.M., CACHOFEIRO V., LAHERA V. Endothelial dysfunction in spontaneously hypertensive rats: consequences of chronic treatment with losartan or captopril. Hypertension. 1997;15:613–618. doi: 10.1097/00004872-199715060-00007. [DOI] [PubMed] [Google Scholar]
- ROSEN G.M., FREEMAN B.A. Detection of superoxide generated by endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7269–7273. doi: 10.1073/pnas.81.23.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGEL G., RÜCHBORN K., SCHNALKE F., GROTE J. Membrane physiological reactions of human arterosclerotic coronary arteries to hypoxia. J. Cardiovasc. Pharmacol. 1992;20:S217–S220. doi: 10.1097/00005344-199204002-00062. [DOI] [PubMed] [Google Scholar]
- SIEGLE I., NÜSING R., BRUGGER R., SPRENGER R., ZECHER R., ULLRICH V. Characterization of monoclonal antibodies generated against bovine and porcine prostacyclin synthase and quantitation of bovine prostacyclin synthase. FEBS Lett. 1994;347:221–225. doi: 10.1016/0014-5793(94)00504-4. [DOI] [PubMed] [Google Scholar]
- SZABO C., SALZMAN A.L., ISCHIROPOULOS H. Endotoxin triggers the expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in the rat aortae in vivo. FEBS Lett. 1995;363:235–238. doi: 10.1016/0014-5793(95)00322-z. [DOI] [PubMed] [Google Scholar]
- TADDEI S., VIRDIS A., MATTEI P., SALVETTI A. Vasodilation to acetylcholine in primary and secondary forms of hypertension. Hypertension. 1993;21:929. doi: 10.1161/01.hyp.21.6.929. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI K., NAMMOUR T.M., FUKUNAGA M., ENBERT J., MORROW J.D., ROBERTS L.J., HOOVER R.L., BADR K.F. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi-prostaglandin F2α, in the rat. Evidence for interaction with thromboxane A2 receptor. J. Clin. Invest. 1992;90:136–141. doi: 10.1172/JCI115826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B. Free radicals in diabetic endothelial cell dysfunction. Free Radical Biol. Med. 1994;16:383–391. doi: 10.1016/0891-5849(94)90040-x. [DOI] [PubMed] [Google Scholar]
- TESFAMARIAM B., COHEN R.A. Role of superoxide anion and endothelium in vasoconstrictor action of prostaglandin endoperoxide. Am. J. Physiol. 1992;262:H1915–H1919. doi: 10.1152/ajpheart.1992.262.6.H1915. [DOI] [PubMed] [Google Scholar]
- TREASURE C.B., KLEIN L., HARRISON D.G., ALEXANDER R.W. Aspirin is associated with improved endothelial vasodilator function in the coronary microvessels of hypertensive patients. Circ. Res. 1991;84:11–135. [Google Scholar]
- VANE J.R., ANGGARD E.E., BOTTING R,M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- VILLA L.M., SALAS E., DARLEY-USMAR V., RADOMSKI M., MOCADA S. Peroxynitrite induces both vasodilation and impaired vasorelaxation in the isolated perfused rat heart. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12383–12387. doi: 10.1073/pnas.91.26.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG P., ZWEIER J.L. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. J. Biol. Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- WHITE C.R., DARLEY-USMAR V., BERRINGTON W.R., MCADAMS M., GORE J.Z., THOMPSON J.A., PARKS D.A., TARPEY M.M., FREEMAN B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIZEMANN T.M., GARDNER C.R., LASKIN J.D., QUINONES S., DURHAM S.K., GOLLE N.L., OHNISHI S.T., LASKIN D.L. Production of nitric oxide and peroxynitrite in the lung during acute endotoxemia. J. Leukoc. Biol. 1994;56:759–768. doi: 10.1002/jlb.56.6.759. [DOI] [PubMed] [Google Scholar]
- ZEIHER A.M., DREXLER H., SAURBIER B., JUST H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J. Clin. Invest. 1993;92:652–662. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZOU M.H., MARTIN C., ULLRICH V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol. Chem. 1997;378:707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- ZOU M.H., ULLRICH V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
- ZOU M.H., KLEIN T., PASQUET J.P., ULLRICH V. Interleukin-1β decreases prostacyclin synthase activity in rat mesangial cells via endogenous peroxynitrite. Biochem. J. 1998;336:507–512. doi: 10.1042/bj3360507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZWEIER J.L., FLAHERTY J.T., WEISFELDT M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]