

Abstract
Peroxisome proliferator-activated receptor (PPAR)s are a family of three nuclear hormone receptors, PPARα, -δ, and -γ, which are members of the steriod receptor superfamily. The first member of the family (PPARα) was originally discovered as the mediator by which a number of xenobiotic drugs cause peroxisome proliferation in the liver. Defined functions for all these receptors, until recently, mainly concerned their ability to regulate energy balance, with PPARα being involved in β-oxidation pathways, and PPARγ in the differentiation of adipocytes. Little is known about the functions of PPARδ, though it is the most ubiquitously expressed. Since their discovery, PPARs have been shown to be expressed in monocytes/macrophages, the heart, vascular smooth muscle cells, endothelial cells, and in atherosclerotic lesions. Furthermore, PPARs can be activated by a vast number of compounds including synthetic drugs, of the clofibrate, and anti-diabetic thiazoldinedione classes, polyunsaturated fatty acids, and a number of eicosanoids, including prostaglandins, lipoxygenase products, and oxidized low density lipoprotein. This review will aim to introduce the field of PPAR nuclear hormone receptors, and discuss the discovery and actions of PPARs in the cardiovascular system, as well as the source of potential ligands.
Keywords: PPAR, macrophage, endothelial cells, vascular smooth muscle, angiogenesis, atherosclerosis
Introduction
The vascular endothelial cell layer plays a critical role in the homeostasis of vascular tissue and blood components. By producing mediators such as prostacyclin, nitric oxide and plasminogen activator, which promote vasodilatation and an anti-thrombotic surface, ECs stop platelets and inflammatory cells adhering and aggregating (see Drexler & Hornig, 1999). Similarly, vascular smooth muscle cells perform important homeostatic functions, providing structural support, and controlling vascular resistance, which ultimately controls blood pressure. If the endothelium or vessels are damaged or become dysfunctional, an inflammatory process can follow. Within the vessel wall itself, cytokines and growth factors are released which activate and modify smooth muscle cells, leading to vascular remodelling. Cytokine-activated smooth muscle cells can become a more synthetic or ‘secretory' cell type, producing a wide variety of mediators (see Ross, 1999). In large blood vessels secretory smooth muscle cells divide faster, and become migratory, leading to medial hyperplasia and intimal thickening. Furthermore, secretory smooth muscle cells can take up large quantities of oxidized lipoproteins. The inflammatory response leads to the recruitment of monocytes within the vessel wall, by a mechanism involving both the upregulation of chemokines, and adhesion molecule expression (see Ross, 1999). Once there, monocytes become phagocytic macrophages, taking up large quantities of oxidized LDL to become lipid laden foam cells (see Ross, 1999; Berliner & Heinecker, 1996).
Peroxisome proliferator-activated receptors (PPARs) are a family of at least three nuclear receptors (α, δ, also referred to as PPARβ FAAR or NUC1 and γ; Issemann & Green, 1990; Dreyer et al., 1992) Until relatively recently their actions were thought limited to specific tissue types, having roles in lipid catabolism, and peroxisome proliferation in the liver (PPARα; Issemann & Green, 1990), and adipogenesis (PPARγ; see Spiegelman & Flier, 1996; see Spiegelman, 1998). Although PPARδ is almost ubiquitously expressed (Kliewer et al., 1994; Braissant et al., 1996; Mukherjee et al., 1997), due to the lack of any selective agonists or antagonists, its roles have yet to be ascertained. For this reason discussion will be mainly limited to PPARα and PPARγ subtypes. Over the last 2 years however it has become apparent that different PPARs are present in a variety of different cell types. The finding that spleen contained relatively high expression of PPARγ mRNA (Kliewer et al., 1994; Braissant et al., 1996), lead to the discovery that monocytes, and especially elicited macrophages, contained PPARγ (Ricote et al., 1998a). Subsequently, different PPARs have been shown to be expressed both in vascular endothelial cells and smooth muscle cells in culture, and in the atherosclerotic lesions. This review aims to discuss this data in the context of what is known about PPARs, and discuss how the modulation of the PPAR pathway may lead to novel therapeutic targets for vascular and other inflammatory diseases.
PPAR ligands
PPARs can be activated by a number of ligands (see Table 1; Issemann & Green, 1990; Yu et al., 1995; Brun et al., 1996; Forman et al., 1997; Kliewer et al., 1997; Nagy et al., 1998), including the fatty acids docosahexaenoic acid, linoleic acid, WY-14643 (selective for PPARα), the anti-diabetic thiazoldinediones (troglitizone, rosiglitizone, and ciglitizone), a number of eicosanoids, including, 5,8,11,14-eicosatetraynoic acid (ETYA), LTB4; the prostanoids PGA1, PGA2, (dehydration products of PGE1 and PGE2 respectively) PGI2, and PGD2. A number of non-steriodal anti-inflammatory drugs also can activate PPARα and PPARγ (Lehmann et al., 1997), albeit at concentrations far exceeding their ability to inhibit COX activity (Mitchell et al., 1993).
Table 1.
The pharmacological activators of PPARs
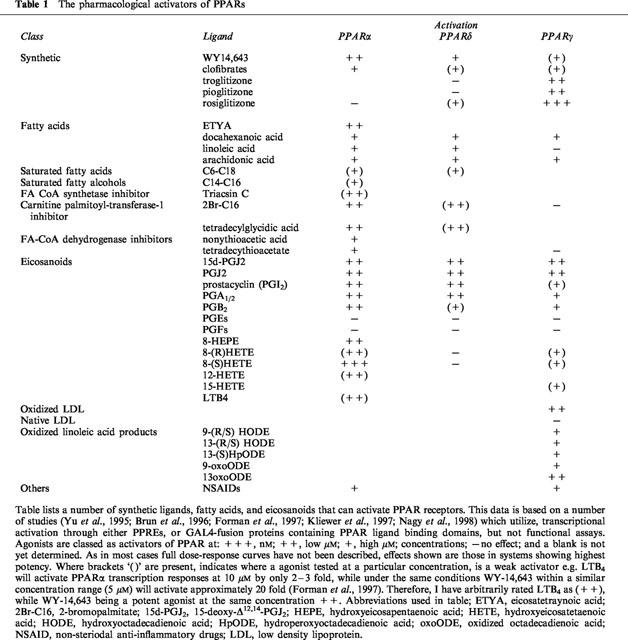
LTB4 was one of the first endogenous ligands described for PPARα. The activation of PPARα by LTB4 causes a negative feedback pathway, whereby the β-oxidation pathway is induced leading to its own degradation (Devchand et al., 1996). Interestingly, the PGD2 dehydration product 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2), was the first endogenous ligand for PPARγ discovered (Forman et al., 1995; Kliewer et al., 1995). Since then, components of oxidized low density lipoprotein (OxLDL), including 9- and 13-hydroxyoctadecadienoic acid (HODE), as well as the further oxidized derivatives have been described as endogenous PPARγ activators (Nagy et al., 1998). 12- and 15-hydroxyeicosatetaenoic acid (HETE) and 13-HODE are also metabolites of 12/15-LOX, utilizing either arachidonic acid (12 or 15-HETE) or linoleic acid (13-HODE) as substrate (see Funk, 1996). The support for 12-, 15-HETE, and 13-HODE, as endogenous PPARγ ligands has recently been greatly strengthened by the finding that 12/15 lipoxygenase activity induced by IL-4 also leads to the release of PPARγ ligands (Huang et al., 1999). As many of these endogenous mediators activate PPARs in the high nM, though most commonly in the low μM range, it is still debatable whether they are the true ‘endogenous' ligands. Furthermore, although these are primarily described as PPARγ ligands, compounds, such as 15d-PGJ2 in some transcription assays are as effective an activator of PPARα or -δ, as they are of PPARγ (Brun et al., 1996). Though, with many of these unstable lipid hydroperoxide mediators the amounts required to activate PPARs by exogenous application, may far exceed those required from endogenous sources. Local production of an agonist may be of sufficient concentration to activate PPARs, while many of the lipids or fatty acids applied exogenously may be metabolized before they come in to proximity with the receptor. One piece of evidence that supports this, is a study of the oestrogen induced peroxisome proliferation mediated by PPARγ in the duck uropygial gland. In this tissue oestrogen induces production of a PPARγ activator, which is a PGD2 metabolite and indistinguishable from Δ12-PGJ2 (Ma et al., 1998). In contrast to this report, NIH3T3 cells which over-express ADD1, the rat homologue of the sterol regulatory element binding protein-1, release a PPARγ agonist which on initial characterization was not 15d-PGJ2 (Kim et al., 1998). The identification of highly potent endogenous ligands for PPARs therefore still remains elusive.
Recently the crystal structure of the ligand binding domains of PPARγ (Nolte et al., 1998; Uppenberg et al., 1998), and PPARδ (Xu et al., 1999) have been solved, revealing a large ligand binding pocket, which has led to the suggestion that maybe PPARs act as general lipid sensors to a broad spectrum of ligands, none of which having a particular high affinity. Since however, many of these eicosanoids and fatty acids are produced and act in the vasculature, or have been discovered in conjuncture with the recent findings demonstrating PPARs in vascular and inflammatory cells, there has been a great deal of interest in these pathways as potential new targets for cardiovascular diseases.
Expression of PPARs and retinoid X receptors
PPARs act as transcription factors upon ligand induced heterodimerization with the common nuclear receptor binding partner, the retinoid X receptor (RXR; Mangelsdorf et al., 1990; see Mangelsdorf & Evans, 1995). Since the original discovery of PPARα from mouse liver (Issemann & Green, 1990), as a nuclear receptor that responded to peroxisome proliferators such as WY-14,643, nafenopin, and clofibric acid, the related family members, PPARδ (PPARβ; Dreyer et al., 1992), and PPARγ (Dreyer et al., 1992) were discovered, and since found to be expressed in a number of species including mice (Issemann & Green, 1990; Zhu et al., 1993; Kliewer et al., 1994), humans (Schmidt et al., 1992; Sher et al., 1993; Mukherjee et al., 1997), xenopus (Dreyer et al., 1992) and rats (Gottlicher et al., 1992). In adult rodents or man, PPARs are differentially expressed (see Table 2; Kliewer et al., 1994; Braissant et al., 1996; Mukherjee et al., 1997). PPARα is found predominantly in the liver, heart, kidney, brown adipose and stomach mucosa. PPARγ is found primarily in adipose tissue, where it plays a critical role in the differentiation of pre-adipocytes into adipocytes, but also in the large intestine, spleen and heart, while PPARδ is almost ubiquitously expressed. RXR also exists in multiple isoforms, RXRα, -β, and -γ (see Chambon, 1996), and like PPARs have a different tissue distribution (see Table 2; Mangelsdorf et al., 1992). It is not known however, if any one of these particular RXR isoforms preferentially bind one or more of the PPAR isoforms.
Table 2.
Relative tissue expression of PPAR and RXR isoforms

Molecular mechanism of the PPARs
RXR isoforms are activated by 9-cis retinoic acid (Kliewer et al., 1992), although other synthetic ‘rexiniod' ligands such as LG268 are also potent specific activators (Boehm et al., 1995). As well as interacting with PPARs, RXR is a common binding partner for a number of other nuclear hormone receptors. RXR can homodimerize, or heterodimerize with thyroid hormone receptor, retinoic acid receptor, vitamin D receptor, as well as the orphan receptors liver X receptor, farsenol X receptor, and the pregnane X receptor (see Mangelsdorf & Evans, 1995). These receptors are activated by different ligands and lead to specific responses. In some combinations of RXR heterodimers (e.g.; RAR: retinoic acid receptor), the RXR ligand does not contribute to activation of a gene by the RXR heterodimer. However, when combined as a PPAR:RXR heterodimer, PPAR ligands and 9-cis retinoic acid can act synergistically on PPAR responses (Kliewer et al., 1992; see Figure 1). The way in which the different dimers of RXR allow specific responses to follow is by binding highly specific sequences in the promoter regions of the various genes they are to trans-activate (see Mangelsdorf & Evans, 1995). The specificity of these dimers in inducing different gene transcription is due to subtle differences in the ‘response element' promoter region they are able to bind. These response elements for nuclear hormone receptors have a general patterns of either direct repeats, palindromic or inverse palindromic sequences separated by one or more nucleotides (see Glass, 1994). For all the PPARs, the PPAR:RXR preferentially binds a direct repeat of the consensus sequence ‘AGGTCA' separated by 1 nucleotide (or a DR1 sequence) called the PPAR response element (PPRE). For example the rat acyl Co oxidase gene which has the first characterized PPRE, has the sequence AGGaCA A AGGTCA (Tugwood et al., 1992). Though, for all the RXR, nuclear receptors heterodimers, each has its own particular affinity to promoter regions of direct repeated sequences spaced by 1–5 nucleotides (called the 1–5 rule; see Mangelsdorf & Evans, 1995).
Figure 1.
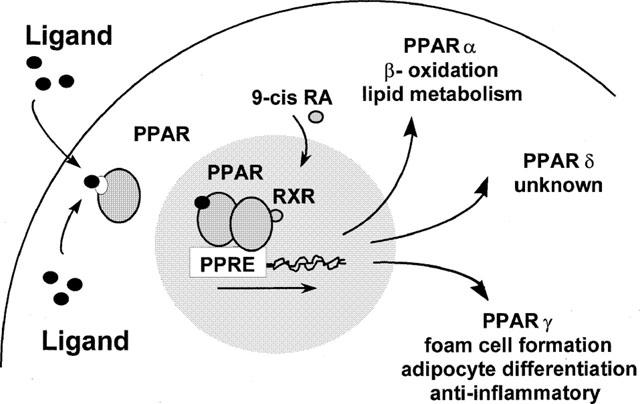
Activation of the PPAR receptors leads to an accumulation in the nucleus, where they heterodimerize with RXR. The PPAR : RXR heterodimer binds to DNA sequences called PPAR response elements (PPRE), leading to the transcription of the responsive gene. Activation of PPARα can cause peroxisome proliferation, and increased lipid metabolism; PPARγ causes cellular lipid accumulation in susceptible cells, and the down regulation of monocyte activation by cytokines; while the function of PPARδ is not known.
Although the PPAR:RXR dimer is the focus for determining specific gene transcription upon ligand activation, transactivation of a particular gene requires a large complex of proteins. Formation and components of such complexes have been reviewed extensively elsewhere (Torchia et al., 1998; Bjorkland et al., 1999). In the un-activated state the PPARs are thought to be in complexes bound with co-repressor proteins such as the silencing mediator for RXR and thyroid receptor or the nuclear receptor co-repressor. Furthermore, at this stage, in some but not all cell types, PPARs may also have a cytoplamsic rather than nuclear sub-cellular location (Chinetti et al., 1998; Bishop-Bailey & Hla, 1999). Upon ligand activation, PPARs dissociated from co-repressors and recruit co-activators, including the PPAR-binding protein (Zhu et al., 1997) and the steroid receptor co-activator-1 (Zhu et al., 1996), and can translocate from the cytoplasm to the nucleus (Bishop-Bailey & Hla, 1999). Indeed, transcription factors act in large complexes, which facilitates their active recruitment to the correct site and orientation within the promoter region of the target gene. Many of the proteins known to be associated in such complexes also contain histone deacetylase activity, to allow for chromatin remodelling and access for the RNA polymerase (see Torchia et al., 1998; Bjorkland et al., 1999).
As the different PPARs have very specific yet diverse actions it is still uncertain how PPARα, -δ and -γ all act on the same DR-1 consensus sequence. Apart from a difference in tissue expression, it is likely that PPARs gain selectivity in part by acting on slight differences in the PPREs from that of the consensus sequence. Indeed, in vitro study of the binding of PPARα, -δ and -γ:RXR heterodimers to PPREs, indicates that although PPARα, -δ and -γ equally bind the rat acyl CoA oxidase PPRE, the PPARγ:RXR heterodimer preferentially binds aP2 (a gene involved in the adipogensis gene activation program) PPREs (Brun et al., 1996). Though this may still be a simplification of how specificity is achieved. Over-expression of PPARδ can inhibit PPARα, or thyroid hormone receptor mediated transcriptional activation, by competition for the RXR binding partner (Jow & Mukherjee, 1995). Futhermore, two high affinity partial agonists for PPARγ have been recently described, MC-555 (Reginato et al., 1998a), and GW0072 (Oberfield et al., 1999). MC-555 and GW0072 are believed to act by virtue of causing a conformational change in the receptor that can only weakly recruit the co-activators; steroid receptor co-activator-1, PPAR binding protein (Oberfield et al., 1999), or cyclic AMP response element binding protein, ‘binding protein' (Reginato et al., 1998a). Apart from being a partial agonist, GW0072, acts to antagonize full agonist (rosiglitizone) induced adipocyte differentiation (Oberfield et al., 1999). A specific response through the PPAR pathway, may therefore be the result of a combination of tissue specificity, competition with other binding partners for the RXR, and the ability of the particular ligand to induce active receptor conformation, and recruit different co-activation complexes. This complexity when fully unravelled will however lead to the possibility of a number of targets for pharmacological or genetic intervention.
PPARs in cardiovascular disease
Over the last few years PPARs have been shown to play an important role in energy homeostasis, diabetes (see Spiegelman, 1998) and obesity (Ristow et al., 1998), cellular differentiation, including considerable inhibitory effects, more recently described on tumour growth (Kubota et al., 1998; Mueller et al., 1998; Brockman et al., 1998; Demetri et al., 1999; Sarraf et al., 1998). For interested readers, the cited reviews, and articles give a good overview of the current published data. In terms of this review, the recent data on the expression of PPARs and potential ligands in vascular, and inflammatory cells will be discussed in more detail.
Monocytes/macrophages
Macrophages are the predominant inflammatory leukocyte in a number of chronic inflammatory diseases including atherosclerosis (see Ross, 1999). In the atherosclerotic lesion they are not only believed to contribute to the inflammation at the level of ‘classical' mediator secretion, but they may also become lipid laden foam cells, and through the release of oxidants, or production of oxidizing enzymes, contribute to the formation of the highly damaging oxidized low density lipoproteins (OxLDL; see Berliner & Heinecke, 1996).
PPARγ is induced in monocytes upon their differentiation in vitro or in vivo to macrophages (Ricote et al., 1998a,1998b). Indeed, isolated monocytes or myeloid precursors activated by PPARγ and RXR ligands, have increased macrophage/monocyte markers CD11b, CD18, and CD14 (Tontonoz et al., 1998). Moreover, monocytes/macrophages treated with PPARγ and RXR ligands increase lipid accumulation and express the OxLDL scavenger receptor CD36 (Tontonoz et al., 1998). PPARγ may therefore be involved in the differentiation from the myeloid precursors, of monocytes, to macrophages, further to foam cells (Tontonoz et al., 1998). LDL particles are a rich potential source of fatty acids, and eicosanoid precursors, which may be potential PPAR ligands. Apart from the apoB core protein, LDL is rich in phospholipids, cholesterol, cholesterol esters, and triglycerides (see Steinberg, 1997). In a closely related article, these authors also demonstrated that OxLDL and the components 9- and 13-HODE, and oxidized derivatives were PPARγ ligands, that induced macrophage foam cell formation (Nagy et al., 1998). Furthermore, similar to the other PPARγ ligands, OxLDL and its components induce the OxLDL scavenger receptor CD36 on macrophages, indicating a positive feedback for lipid accumulation (Nagy et al., 1998), potentially explaining the ability of PPARγ to induce foam cell formation. In slight contrast, in activated macrophages, the stimulation of PPARγ by ligands also results in a global inhibition of inflammatory mediator production from these cells. PPARγ ligands inhibit IFN-γ induced morphogenesis, inducible nitric oxide synthase induction, gelatinase B and scavenger receptor-A expression (Ricote et al., 1998a), and the release of the pro-inflammatory cytokines TNF-α, IL-6, IL-1β and IL-2 (Jiang et al., 1998). Further analysis revealed that PPARγ can actually trans-repress the activity of these inflammatory mediators at the level of transcription, by inhibiting nuclear factor (NF)-κB, Stat1, and activation protein-1 signalling (Ricote et al., 1998a); some of the central transcription factors implied in inflammatory responses.
In differentiated macrophages, at least in vitro, PPARs have yet a different function, in that both PPARα and -γ ligands induce macrophage apoptosis (Chinetti et al., 1998). Little is known about how PPARs act in other inflammatory leukocytes, though murine T-helper1 lymphocytes express at least PPARγ, activation of which leads to the inhibition of antigen or T-cell receptor induced proliferation (Clark et al., 1999). Human neutrophils, and peripheral blood lymphocytes express a truncated PPARγ message, which does not fully code the protein (Greene et al., 1995).
PPARγ and -α are clearly expressed in monocytes/ macophages in vitro. In vivo a high expression of PPARγ is also observed in experimental (Tontonoz et al., 1998) and human atherosclerotic lesions (Ricote et al., 1998b; Marx et al., 1998b) predominantly in the macrophages, but also to the endothelial cell layer, and the vascular smooth muscle (more strongly in the intimal than medial layer). PPARα and PPARδ are also expressed in atherosclerotic lesions (D Bishop-Bailey, E Smith, C Haudenschild and T Hla, unpublished observations). PPARα has a similar expression pattern to PPARγ, while PPARδ expression is much weaker, being sparsely expressed in the endothelial cell layer, intimal but not medial smooth muscle cells, and in adherent or infiltrated cells, presumably monocytes or macrophages.
Previous findings in the macrophage would appear to suggest opposing sides of PPARγ activation, an anti-inflammatory effect, and a pro-atherogenic effect. Which of these many functions ascribed to PPARs in monocytes/ macrophages predominates in vivo is yet to be ascertained. Though the effects subsequently demonstrated in vascular smooth muscle cells and endothelial cells, again show multiple sometimes opposing actions.
Vascular smooth muscle
Vascular smooth muscle contains PPARα (Staels et al., 1998a; Marx et al., 1998a), PPARγ (Ricote et al., 1998b; Marx et al., 1998a) and PPARδ (D Bishop-Bailey, unpublished observations in rat aortic vascular smooth muscle cells, by Northen blot analysis, and immunofluorescence). Before the realization of the existence of PPARs in vascular smooth muscle cells, and that thiazoldinediones were potent PPARγ activators, troglitizone was shown to inhibit vascular smooth muscle proliferation and migration in vitro, and restenosis in a model of balloon angioplasty in vivo (Law et al., 1996). Indeed, in this study troglitizone also inhibited c-fos induction, and serum response element induced transcriptional activation. These effects of troglitizone are presumably through PPARγ, though direct effects on Ca2+ channels have also been noted (Zhang et al., 1994; Song et al., 1997). Subsequently, PPARγ expression was demonstrated in human vascular smooth muscle cells, and PPARγ, but not PPARα agonists, inhibited platelet derived growth factor-BB induced migration (Marx et al., 1998a). One of the mechanisms required for cellular migration is the degradation of extracellular matrix. Similar to macrophages (Ricote et al., 1998a), troglitizone, and 15d-PGJ2 selectively inhibited phorbol ester induced vascular smooth muscle cell expression of gelatinase-B (matrix metalloproteinase-9). The expression of matrix metalloproteinase-2, or tissue inhibitors of matrix metalloproteinase-1 or -2 was unaffected (Marx et al., 1998a).
In contrast PPARα, but not PPARγ ligands, inhibit inflammatory responses in vascular smooth muscle cells by repressing NF-κB signalling (Staels et al., 1998a). Indeed, by this mechanism, fenofibrate, and WY-16,463 inhibits IL-1 induced IL-6 production, and inducible cyclo-oxygenase (COX-2) expression (Staels et al., 1998a). These results are consistent with findings in the PPARα knockout mouse which has increased inflammatory responses, though this is considered in part due to the reduced capacity to metabolize inflammatory mediators through β-oxidation (Devchand et al., 1996). Moreover, hyperlipidaemic patients treated with fenofibrate also had reduced circulating IL-6 and acute phase proteins (Staels et al., 1998a). Although, it is unclear if fenofibrate is exclusively a PPARα agonist in vivo, a number of beneficial cardiovascular effects may result from PPARα activation in the liver, which is the central organ regulating lipid metabolism and lipoprotein expression. As recently reviewed by Staels et al. (1998b) PPARα activation in the liver by the fibrate class of drugs leads to the increase in protective high density lipoproteins, while decreasing detrimental LDLs and VLDL synthesis, enhances lipolysis, and increases fatty acid catabolism.
Both PPARα and -γ appear to have protective effects on the activity of vascular smooth muscle cells. PPARα ligands, reduce the inflammatory response within the vessel wall by at least interfering with NF-κB signalling, while PPARγ ligands seem to specifically inhibit smooth muscle migration, by interfering with processes involved in the degradation of the extracellular matrix.
Vascular endothelial cells
Vascular endothelial cells contribute to the release of protective mediator in large blood vessels, where endothelial damage or dysfunction is considered one of the potential mechanisms for development of atherosclerosis and restenosis (see Drexler & Hornig, 1999). In the microvasculature, the endothelium contributes to chronic inflammatory processes, wound healing, and tumour formation by an angiogenic response, vascularizing the newly produced tissue (Folkman, 1995). Vascular endothelial cells contain PPARα (Inoue et al., 1998; Xin et al., 1999; Bishop-Bailey & Hla, 1999), PPARδ (Xin et al., 1999; Bishop-Bailey & Hla, 1999), and PPARγ (Marx et al., 1999a; Xin et al., 1999; Bishop-Bailey & Hla, 1999), though, as yet no definitive roles for PPARδ have been described. PPARα agonists, similar to their effects in vascular smooth muscle cells inhibit NF-κB signalling, by which mechanism they can inhibit TNF-α induced expression of vascular cell adhesion molecule-1, and the subsequent adherence of monocytes (Marx et al., 1999b). Like other cell types PPARγ appears to have both potential protective and detrimental effects. In terms of large vessel disease, PPARγ activators inhibit the endothelial cell release of endothelin-1 (Satoh et al., 1999; Delerive et al., 1999), a potent vascoconstrictor, and vascular smooth muscle cell mitogen (see Ruschitzka et al., 1997). In contrast, PPARγ agonists also increase plasminogen activator inhibitor (PAI)-1 expression (Marx et al., 1999a; Xin et al., 1999). PAI-1 is highly expressed in adipocytes, and levels correlate with obesity, furthermore, increased PAI-1 is associated with myocardial ischaemia, and thrombosis (Loskutoff & Samad, 1997). A role which is unsurprising since PAI-1 inhibits two of the enzymes which play a major role in fibrinolysis, tissue plasminogen activator and urokinase. The reverse side to the inhibition of PAI-1 in large vessels, is that in small vessels increasing PAI-1 (Xin et al., 1999) may be one of the mechanisms responsible for inhibition of angiogenesis by PPARγ agonists (Xin et al., 1999; Bishop-Bailey & Hla, 1999). Furthermore, PPARγ agonists also reduce the vascular endothelial cell growth factor receptors Flt-1, and Flk/KDR, and inhibit the expression of urokinase (Xin et al., 1999). Alternatively, PPARγ activators can inhibit endothelial cell angiogenesis by inducing apoptosis (Bishop-Bailey & Hla, 1999), similar to macrophages (Chinetti et al., 1998), via a caspase-3 mediated process. Though again in contrast to the microvasculature, in a large vessel disease, inappropriate apoptosis may cause structural weakness in an atherosclerotic lesion, and may promote plaque rupture (Newby & Zaltsman, 1999), which may lead to embolism or stroke.
In the microvasculature PPARγ may be a novel target for anti-angiogenic therapy for a number of tumours or chronic inflammatory disorders. Like other cells, PPARγ activation can inhibit cytokine induced mediator release, which may be beneficial in large vessel disease. Many of the effects described which would be beneficial in the microvasculature however, inhibiting endothelial cell function, may naturally be detrimental to protecting against atherogenic stimuli.
The heart
Although, the heart contains very high levels of all PPAR and RXR receptors (see Table 2), very little is known about how PPAR ligands effect cardiac function. Certainly PPARα appears to have an important role in mitochondrial fatty acid β-oxidation (Brandt et al., 1998; Yu et al., 1998). Indeed, in PPARα knockout mice, once mitochondrial fatty acid import is blocked by the metabolic inhibitor etomoxir, the mice undergo a hypoglycaemia, and a massive increase in lipid accumulation in the heart and liver, resulting in 100% mortality of male mice (Djouadi et al., 1998); females being largely (75%) protected by oestrogens (Djouadi et al., 1998). Nothing however is known with regards to the potential role of PPARs in the pathologies of the heart, or cardiac tissue.
Potential sources of PPAR ligands in the cardiovascular system
Although PPARs are present in many different cell types, and are activated by a large variety of ligands, very little is known about the ‘endogenous' pathways for PPAR activation. The homeostasis of the vasculature and the pathogenesis of vascular diseases such as atherosclerosis involves regulation of fatty acid metabolism, the uptake and accumulation of oxidized lipids, and the induction of inflammatory responses. With so many PPAR ligands now discovered (see Table 2), there are several potential pathways to form these agonists. While fatty acids are essential ubiquitous components of cell systems, other endogenous pathways for PPAR ligand production may be associated with other ‘house-keeping' roles of cells, or subsequent disease processes. Indeed, vascular and inflammatory cells express, and can be induced to express cyclo-oxygenase and lipoxygenase enzymes, and many diseases processes are associated with increases in oxidized lipids.
Cyclo-oxygenases
A number of COX products are PPAR agonists (see Table 2), including 15d-PGJ2, a dehydration product of PGD2, as well as PGI2, one of the major products of vascular COXs (see Bishop-Bailey et al., 1999). Constitutive COX-1 is abundant in endothelial cells, while COX-2 can be readily induced in human endothelial cells (Hla & Nielson, 1992) and smooth muscle cells (Bishop-Bailey et al., 1997; 1998) to produce large quantities of prostanoids, of which PGI2 and PGE2 have protective vascular functions (see Bishop-Bailey et al., 1999). Similarly, monocytes/and macrophages express low levels of COX-1, and can also be readily induced to express high levels of COX-2 and produce large quantities of prostanoids (O'Banion et al., 1992). Moreover, COX-2 is highly expressed in human atherosclerotic lesions (Baker et al., 1999), and both COX-1 (Narko et al., 1997) and COX-2 (Tsujii et al., 1998) have been implicated in tumour formation, and angiogenesis. The pattern of prostanoid release depends on the presence of secondary metabolizing enzymes. However in the absence of secondary enzymes, PGF2α, PGD2 and in particular PGE2 can be formed non-enzymically (see Smith et al., 1991). Vascular endothelial cells, and vascular smooth muscle contain large amounts of PGI2 synthetase (Smith et al., 1983), though in atherosclerotic lesions, synthetic smooth muscle cells also express the ‘neuronal' lipocalin type PGD synthase (Eguchi et al., 1997). Although, COX preferentially metabolizes arachidonic acid, it can also use a number of other free fatty acids such as 8,11,14-eicosatrieoic acid and linoleic acid as alternative substrates. For this reason COX can also form, under certain conditions, members of other biologically active lipid families, 11-, and 15-HETE; 9- and 13-HODE; 12-hydroxyheptadeatrienoic acid; and isoprostanes (see Bishop-Bailey et al., 1999). Interestingly, a number of COX products which can activate the MAP kinase pathways, such as PGF2α may also indirectly regulate PPAR pathways at the level of receptor phosphorylation (Reginato et al., 1998b).
Lipoxygenase
LOXs are a large family of enzymes which can metabolize mainly arachidonic acid, to a number of biologically active mediators (see Funk et al., 1996); a number of which have since been demonstrated to activate PPARs. The three main classes of LOXs are those with 5-LOX, 12-LOX, or 15-LOX activity, which are expressed in a variety of primarily inflammatory cell types, platelets, epidermal tissue (see Funk et al., 1996), and cardiomyocytes (Breibart et al., 1996). 5-LOX metabolize arachidonic acid to 5-HPETE, the precursor for LTs (LTB4 of which is a PPARα agonist), 12-LOX (rodent homologue of 15-LOX) metabolizes arachidonic acid to 12-HPETE>>15-HPETE, while 15-LOX conversely metabolizes arachidonic acid to 15-HPETE>>12-HPETE, precursors for their respective HETE moieties (see Funk, 1996). Arachidonic acid is not however the only substrate for 12/15-LOX, as both linoleic acid (metabolized to 13(S)HODE), and cholestryl linoleate, one of the major lipid components of LDL can be utilized. Furthermore, it has also been suggested that LOX activity, may initiate oxidization of LDL, into toxic OxLDL (see Steinberg, 1997). 15-LOX is found upregulated, and co-localized to lipid rich atherosclerotic lesions (Yla-Herttuala et al., 1990; 1991), both in the macrophage, and in the endothelial cell layer. Murine fibroblasts which over-express 15-LOX enhance lipoperoxides production with LDL as substrate (Benz et al., 1995). Furthermore, apoE/ 12/15-LOX double knockout mice have reduced lesion formation (Cyrus et al., 1999), compared to apoE knockouts alone. Though, LOXs can produce PPAR ligands in vitro (Huang et al., 1999), and are strongly implicated in the progression of a number of diseases, including atherosclerosis, it is still uncertain if these 5-, 12, or 15-LOX products serve as functional PPAR ligands in these processes.
Oxidized LDL and derivatives
The atherosclerotic lesion is known to contain large amounts of oxidized esterified lipids, from OxLDL, 13-HODE (see Feinmark & Cornicelli, 1997), to 5-, 8-, 9-, 11-, 12- and 15-HETEs, 5,6-, 8,9-, 11,12-, and 14,15-epoxyeicosatetraenoic acids, F series isoprostanes (Mallat et al., 1999). Sources of endogenous ligands LDL oxidized derivatives. It is as yet uncertain, how much of these lipids like 13-HODE are produced by enzymatic processes, 15-LOX produces specifically 13(S)-HODE, while 13-HODE found in lesions is primarily racemic, indicative of a non-enzymic mechanism. Such non-enzymic pathways include, oxidation by superoxide anion, by free Fe2+ or Cu2+ ions, or by the reactive intermediates of hydroperoxides, such as 15-HETE formed by LOXs (see Sigal et al., 1994). Thereby, enzymes such as 15-LOX, myeloperoxidase or even cyclo-oxygenase can still contribute indirectly to lipid peroxidation, as opposed to directly utilizing LDL as substrate, by the mediators it releases from the cell. As 15-LOX is intracellular, while lipids largely remain extracellular indeed, this may be the primary mechanism by which 15-LOX causes oxidization of lipids within a lesion.
Conclusion
The early descriptions of PPARs limited their activity to specialist functions, such as β-oxidation and peroxisome proliferation in the liver (PPARα), and adipogenesis (PPARγ) in specialist cell types. It now appears that PPARs, are more widespread, albeit it is often at lower levels in vascular, and inflammatory cells. Furthermore, their actions apart from their classical roles in β-oxidation, and adipogenesis, seemed to have extended, where they now have important contributions in the control of inflammatory responses, cell growth and differentiation. Although the actions of PPARα and PPARγ are becoming clearer, it is still uncertain what roles PPARδ has, especially in the vasculature. Though this may become clearer as PPARδ selective ligands are developed and become available. Furthermore, PPARγ seem to have a number of conflicting actions within inflammatory and vascular cells, appearing to both be able to have anti-inflammatory actions, as well as inducing foam cell formation, and causing apoptosis. Which of these roles dominates in vivo is still unsure, though very likely to be solved in the near future with the use of the potent selective agonists now available. Since now the crystal structures for PPARγ and PPARδ have been solved, it should help to progress the development further of selective agonists, and may certainly help the development of antagonists for these receptors. PPARs seem to be able to respond to a vast number of ligands, from fatty acids, to eicosanoid and linoleic acid metabolites. One of the major prospectives in this field may be to find if these receptors are general lipid sensors, or in fact there are as yet unidentified highly potent endogenous ligands. PPARs have now been demonstrated in a number of diseases, including atherosclerosis, diabetes, inflammation and certain cancers, where the use of selective ligands has often led to beneficial effects. The newly discovered existence of PPARs in vascular and inflammatory cells, opens up the possibility that modulation of these nuclear hormone receptors in the cardiovascular system may have a large therapeutic potential.
Figure 2.
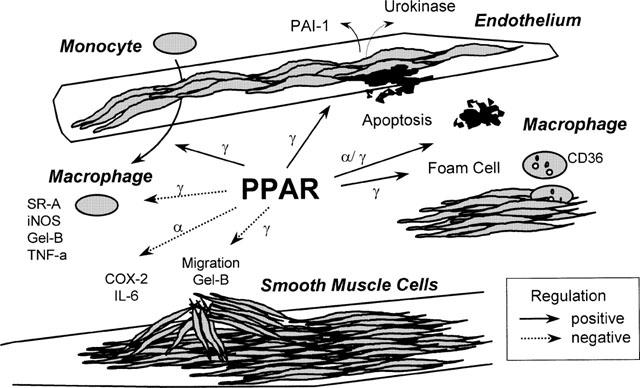
PPARα and PPARγ regulate vascular and inflammatory cell functions. Many different, often contradictory effects have been ascribed to PPAR ligands in vascular, and inflammatory cells. PPARα, and γ are expressed in endothelial cells, monocytes/macrophages, and in the vascular smooth muscle cells of both medial and intimal layers. PPARα ligands inhibit smooth muscle cell production of inflammatory products, and causes macrophage apoptosis. PPARγ ligands (i) inhibit smooth muscle cell migration processes; (ii) inhibit monocyte/macrophage production of inflammatory enzymes (iNOS; inducible nitric oxide synthase; Gel-B, gelatinase B), cytokines, and scavenger receptor (SR)-A expression; (iii) induce monocyte/macrophages differentiation, and uptake of oxidized LDL; (iv) induce monocyte and endothelial cell apoptosis; (v), inhibit the expression of vascular endothelial cell growth factor receptors, endothelin-1 and urokinase expression in endothelial cells and, (vi) induce plasminogen activator inhibitor-1 expression in endothelial cells.
Figure 3.
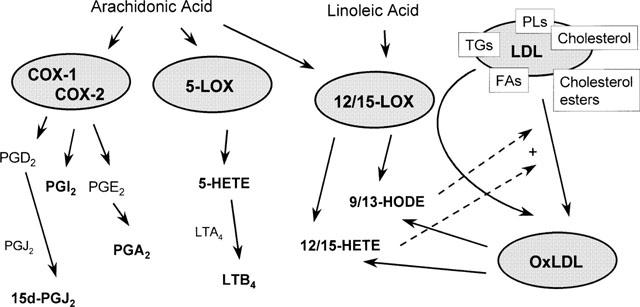
PPAR ligands as products of cyclo-oxygenase (COX), lipoxygenase (LO), or oxidized low density lipoprotein (oxLDL). Little is actually known about which mediators act as true endogenous ligands, therefore the figure describes some of the potential pathways for the production of PPAR ligands. Directly binding PPAR ligands are noted in bold text. Constituitive (COX-1), or inducible (COX-2), present in vascular and inflammatory cells, can utilize primarily arachidonic acid to form PGH2. PGH2 is metabolized further to a number of prostanoid mediators, some of which, 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2, a dehydration product of PGD2), PGA2 (dehydration product of PGE2), and PGI2 (prostacyclin) that activate PPAR receptors. 5-LO, present in leukocytes, forms from arachidonic acid 5-HpETE, the precursor for 5-HETE, which itself is the precursor for leukotrienes (LT)s, of which LTB4 is also a PPAR(α) agonist. 12/15-LOs, whose presence is strongly implied in the pathogenesis of atherosclerosis can produce PPAR agonists from arachidonic acid, the 12- and 15-HETEs, and form linoleic acid, 9- and 13-HODE. Low density lipoprotein LDL, which contains a number of phospholipids (PL)s, triglycerides (TG)s, cholesterol, cholesterol esters, and fatty acids, can be directly oxidized by 12/15-LOs, to produce PPAR ligands including 9- and 13-HODE. Furthermore, these unstable lipid hydroperoxides, 9- and 13-HODE, and the 12- and 15-HETEs, may themselves directly contribute to oxidization of LDL through a non-enzymatic pathway.
Acknowledgments
D. Bishop-Bailey currently holds a British Heart Foundation Intermediate Fellowship. Many thanks to Dr Timothy Hla and Mr Philippe Delerive for useful discussions, and to Dr Nicolas Ancellin and Dr Robert Clark for their critical reading of the manuscript.
Abbreviations
- COX-1
constitutive COX
- COX-2
inducible COX
- 15d-PGJ2
15-deoxy-Δ12,14-PGJ2
- ETYA
eicosatetraynoic acid
- HETE
hydroxyeicosatetaenoic acid
- HODE
hydroxyoctadecadienoic acid
- HPETE
hydroperoxyeicosatetaenoic acid
- LDL
low density lipoprotein
- NF-κB
nuclear factor-κB
- OxLDL
oxidized LDL
- PAI-1
plasminogen activator inhibitor-1
- PPAR
peroxisome proliferator-activated receptor
- PPRE
PPAR response element
- RXR
retinoid X receptor
References
- AUBOEUF D., RIEUSSET J., FAJAS L., VALLIER P., FRERING V., RIOU J.P., STAELS B., AUWERX J., LAVILLE M., VIDAL H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46:1319–1327. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- BAKER C.S., HALL R.J., EVANS T.J., POMERANCE A., MACLOUF J., CREMINON C., YACOUB M.H., POLAK J.M. Cyclooxygenase-2 is widely expressed in atherosclerotic lesions affecting native and transplanted human coronary arteries and colocalizes with inducible nitric oxide synthase and nitrotyrosine particularly in macrophages. Arteriscler. Thromb. Vasc. Biol. 1999;19:646–655. doi: 10.1161/01.atv.19.3.646. [DOI] [PubMed] [Google Scholar]
- BENZ D.J., MOL M., EZAKI M., MORI-ITO N., ZELAN I., MIYANOHARA A., FRIEDMAN T., PARTHASARATHY S., STEINBERG D., WITZTUM J.L. Enhanced levels of lipoperoxides in low density lipoprotein incubated with murine fibroblasts expressing high levels of human 15-lipoxygenase. J. Biol. Chem. 1995;270:5191–5197. doi: 10.1074/jbc.270.10.5191. [DOI] [PubMed] [Google Scholar]
- BERLINER J.A., HEINECKE J.W. The role of oxidized lipoproteins in atherogenesis. Free Radical Biol. Med. 1996;20:707–727. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., HLA T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Δ12,14 prostaglandin J2. J. Biol. Chem. 1999;274:17042–17048. doi: 10.1074/jbc.274.24.17042. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., HLA T., MITCHELL J.A. Cyclo-oxygenase-2 in vascular smooth muscle. Int. J. Mol. Med. 1999;3:41–48. doi: 10.3892/ijmm.3.1.41. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., LARKIN S.W., PEPPER J.R., MITCHELL J.A. Differential induction of cyclo-oxygenase-2 human venous and arterial vascular smooth muscle cells: role of endogenous prostanoids. Arterioscler. Thromb. Vasc. Biol. 1998;18:1655–1661. doi: 10.1161/01.atv.18.10.1655. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., PEPPER J.R., HADDAD E-L., NEWTON R., LARKIN S.W., MITCHELL J.A. Induction of cyclo-oxygenase-2 in human saphenous vein and internal mammary artery. Arterioscler. Thromb. Vasc. Biol. 1997;17:1644–1648. doi: 10.1161/01.atv.17.9.1644. [DOI] [PubMed] [Google Scholar]
- BJORKLUND S., ALMOUZNI G., DAVIDSON I., NIGHTINGALE K.P., WEISS K. Global transcription regulators of eukaryotes. Cell. 1999;96:759–767. doi: 10.1016/s0092-8674(00)80586-3. [DOI] [PubMed] [Google Scholar]
- BOEHM M., ZHANG L., ZHI L., MCCLURG M., BERGER E., WAGONER M., MAIS D., SUTO C., DAVIES J., HEYMAN R., NADZAN A.M. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J. Med. Chem. 1995;38:3146–3155. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- BRAISSANT O., FOUFELLE F., SCOTTO C., DAUCA M., WAHLI W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue ditribution of PPARα, -β, and -γ in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- BRANDT J.M., DJOUADI F., KELLY D.P. Fatty acids activate transcription of the muscle arnitine palmitoyltransferase I gene in cardiac myocytes via peroxisome proliferator-activated receptor α. J. Biol. Chem. 1998;273:23786–23792. doi: 10.1074/jbc.273.37.23786. [DOI] [PubMed] [Google Scholar]
- BREITBART E., SOFER Y., SHAINBERG A., GROSSBERG S. Lipoxygenase activity in heart cells. FEBS Lett. 1996;395:148–152. doi: 10.1016/0014-5793(96)01017-4. [DOI] [PubMed] [Google Scholar]
- BROCKMAN J.A., GUPAT R.A., DUBOIS R.N. Activation of PPARγ leads to inhibtion of anchorage-independent growth of human colorectal cancer cells. Gastroenterology. 1998;115:1049–1055. doi: 10.1016/s0016-5085(98)70072-1. [DOI] [PubMed] [Google Scholar]
- BRUN R.P., TONTONOZ P., FORMAN B.M., ELLIS R., CHEN J., EVANS R.M., SPIEGELMAN B.M. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev. 1996;10:974–984. doi: 10.1101/gad.10.8.974. [DOI] [PubMed] [Google Scholar]
- CHAMBON P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- CHINETTI G., GRIGLIO S., ANTONUCCI M., TORRA I.P., DELERIVE P., MAJD Z., FRUCHART J-C., CHAPMAN J., NAJIB J., STAELS B. Activation of peroxisome proliferator-activated receptor α and γ induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998;273:25573–25580. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- CLARK R.B., BISHOP-BAILEY D., ESTRADA-HERNANDEZ T., HLA T., PUDDINGTON L., PADULA S.J.The nuclear receptor PPARγ and immunoregulation: PPARγ mediates inhibition of helper T cell responses J. Immunol. 1999. In press [DOI] [PubMed]
- CYRUS T., WITZTUM J.L., RADER D.J., TANGIRALA R., FAZIO S., LINTON M.F., FUNK C. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo-E deficient mice. J. Clin. Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELERIVE P., MARTIN-NIZARD F., CHINETTI G., TROTTEIN F., FRUCHART J.C., NAJIB J., DURIEZ P., STAELS B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signalling pathway. Circ. Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- DEMETRI G.D., FLETCHER C.D.M., MUELLER E., SARRAF P., NAUJOKS R., CAMPBELL N., SPIEGELMAN B.M., SINGER S. Induction of solid tumour differentiation by the peroxisome proliferator-activated receptor γ ligand troglitizone in patients with liposarcoma. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVCHAND P.R., KELLER H., PETERS J.M., VAZQUEZ M., GONZALEZ F.J., WAHLI W. The PPARα- leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- DJOUADI F., WEINHEIMER C.J., SAFFITZ J.E., PITCHFORD C., BASTIN J., GONZALEZ F.J., KELLY D.P. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor α-deficient mice. J. Clin. Invest. 1998;102:1083–1091. doi: 10.1172/JCI3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DREXLER H., HORNIG B. Endothelial dysfunction in human disease. J. Mol. Cell. Cardiol. 1999;31:51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- DREYER C., KREY G., KELLER H., GIVEL F., HELFTENBEIN G., WAHLI W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- EGUCHI Y., EGUICHI N., ODA H., SEIKI K., KIJIMA Y., MATSU-URA Y., URADE Y., HAYAISHI O. Expression of lipocalin-type prostaglandin D synthase (β-trace) in human hearts and its accumulation in the coronary circulation of angina patients. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14689–14694. doi: 10.1073/pnas.94.26.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEINMARK S.J., CORNICELLI J.A. Is there a role for 15-lipoxygenase in atherogenesis. Biochem. Pharmacol. 1997;54:953–959. doi: 10.1016/s0006-2952(97)00135-4. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J. Angiogenesis in cancer, vascular rheumatoid and other disease. Nature Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- FORMAN B.M., CHEN J., EVANS R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORMAN B.M., TONTONOZ P., CHEN J., BRUN R.P., SPIEGELMAN B.M., EVANS R.M. 15-Deoxy-Δ-12,14- prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- FUNK C.D. The molecular biology of mammalian lipoxygenases and the quest for eicosanoid functions using lipoxygenase deficient mice. Biochim. Biophys. Acta. 1996;1304:65–84. doi: 10.1016/s0005-2760(96)00107-5. [DOI] [PubMed] [Google Scholar]
- GLASS C.K. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocrinol. Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- GOTTLICHER M., WIDMARK E., LI Q., GUSTAFSSON J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREENE M.E., BLUMBERG B., MCBRIDE O.W., YI H.F., KRONQUIST K., KWAN K., HSIEH L., GREENE G., NIMER S.D. Isolation of the human peroxisome proliferator-activated receptor γ cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Exp. 1995;4:281–299. [PMC free article] [PubMed] [Google Scholar]
- HLA T., NIELSON K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG J.T., WELCH J.S., RICOTE M., BINDER C.J., WILLSON T.M., KELLY C., WITZTUM J.L., FUNK C.D., CONRAD D., GLASS C.K. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- INOUE I., SHINO K., NOJI S., AWATA T., KATAYAMA S. Expression of peroxisome proliferator-activated receptor α (PPARα) in primary cultures of human vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998;246:370–374. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- ISSEMANN I., GREEN S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- JIANG C., TING A.T., SEED B. PPARγ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- JOW L., MUKHERJEE R. The human peroxisome proliferator-activated receptor (PPAR) subtype NUC1 represses the activation of hPPARα and thyroid hormone receptors. J. Biol. Chem. 1995;270:3836–3840. doi: 10.1074/jbc.270.8.3836. [DOI] [PubMed] [Google Scholar]
- KIM J.B., WRIGHT H.M., WRIGHT M., SPIEGELMAN B. ADD1/SREBP1 activates PPARγ through the production of endogenous ligand. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLIEWER S.A., FORMAN B.M., BLUMBERG B., ONG E.S., BORGMEYER U., MANGELSDORF D.J., UMESONO K., EVANS R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLIEWER S.A., LENHARD J.M., WILLSON T.M., PATEL I., MORRIS D.C., LEHMANN J.M. A prostaglandin J metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- KLIEWER S.A., SUNDSETH S.S., JONES S.A., BROWN P.J., WISELY G.B., KOBLE C.S., DEVCHAND P., WAHLI W., WILLSON T.M., LENHARD J.M., LEHMAN J.M. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLIEWER S.A., UMESONO K., NOONAN D.J., HEYMAN R.A., EVANS R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUBOTA T., KOSHIZUKA K., WILLIAMSON E.A., ASOU H., SAID J.W., HOLDEN S., MIYOSHI I., KOEFFLER H.P. Ligand for peroxisome proliferator-activated receptor γ (troglitizone) has potent antitumour effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998;58:3344–3352. [PubMed] [Google Scholar]
- LAW R.E., MEEHAN W., XI X-P., GRAF K., WUTHRICH D.A., COATS W., FAXON D., HSUEH W.A. Troglitizone inhibits vascular smooth muscle cell growth and intimal hyperplasia. J. Clin. Invest. 1996;98:1897–1905. doi: 10.1172/JCI118991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEHMANN J.M., LENHARD J.M., OLIVER B.B., RINGOLD G.M., KLIEWER S.A. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steriodal antiinflammatory drugs. J. Biol. Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- LOSKUTOFF D.J., SAMAD F. The adipocyte and hemostatic balance in obesity: studies of PAI-1. Arteriscler. Thromb, Vasc. Biol. 1997;18:1–6. doi: 10.1161/01.atv.18.1.1. [DOI] [PubMed] [Google Scholar]
- MA H., SPRECHER H.W., KOLATTUKUDY P.E. Estrogen-induced production of a peroxisome proliferator-activated receptor (PPAR) ligand in a PPARγ-expressing tissue. J. Biol. Chem. 1998;273:30131–30138. doi: 10.1074/jbc.273.46.30131. [DOI] [PubMed] [Google Scholar]
- MALLAT Z., NAKAMURA T., OHAN J., LESECHE G., TEDGUI A., MACLOUF J., MURPHY R.C. The relationship of hydroxyeicosatetraenoic acids and F2-isoprostanes to plaque instability in human carotid atherosclerosis. J. Clin. Invest. 1999;103:421–427. doi: 10.1172/JCI3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANGELSDORF D.J., BORGMEYER U., HEYMAN R.A., ZHOU J.Y., ONG E.S., ORO A.E., KAKIZUKA A., EVANS R.M. Characterisation of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- MANGELSDORF D.J., EVANS R.M. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- MANGELSDORF D.J., ONG E.S., DYCK J.A., EVANS R.M. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature. 1990;345:224–229. doi: 10.1038/345224a0. [DOI] [PubMed] [Google Scholar]
- MARX N., BOURCIER T., SUKHOVA G.K., LIBBY P., PLUTZKY J. PPARγ activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression. Arterioscler. Thromb. Vasc. Biol. 1999a;19:546–551. doi: 10.1161/01.atv.19.3.546. [DOI] [PubMed] [Google Scholar]
- MARX N., SCHONBECK U., LAZAR M.A., LIBBY P., PLUTZKY J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ. Res. 1998a;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARX N., SUKHOVA G.K., COLLINS T., LIBBY P., PLUTZKY J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999b;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARX N., SUKHOVA G., MURPHY C., LIBBY P., PLUTZKY J. Macrophages in human atheroma contain PPARγ. Am. J. Pathol. 1998b;153:17–23. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.A., AKARASEREENONT P., THIEMERMANN C., FLOWER R.J., VANE J.R. Selectivity of nonsteriodal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11693–11697. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUELLER E., SARRAF P., TONTONOZ P., EVANS R.M., MARTIN K.J., ZHANG M., FLETCHER C., SINGER S., SPIEGELMAN B.M. Terminal differentiation of human breast cancer through PPARγ. Mol. Cell. 1998;1:465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- MUKHERJEE R., JOW L., CROSTON G.E., PATERNITI J.R. Identification, characterisation, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARγ2 versus PPARγ1 and activation with retinoid X receptor agonists and antagonists. J. Biol. Chem. 1997;272:8071–8076. doi: 10.1074/jbc.272.12.8071. [DOI] [PubMed] [Google Scholar]
- NAGY L., TONTONOZ P., ALVAREZ J.G.A., CHEN H., EVANS R.M. Oxidised LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- NARKO K., RISTIMAKI A., MACPHEE M., SMITH E., HAUDENSCHILD C.C., HLA T. Tumorigenic transformation of immortalized ECV endothelial cells by cyclooxygenase-1 overexpression. J. Biol. Chem. 1997;272:21455–21460. doi: 10.1074/jbc.272.34.21455. [DOI] [PubMed] [Google Scholar]
- NEWBY A.C., ZALTSMAN A.B. Fibrous cap formation or destruction–the critical importance of vascular smooth muscle cell proliferation, migration, and matrix formation. Cardiovasc. Res. 1999;41:345–360. [PubMed] [Google Scholar]
- NOLTE R.T., WISELY G.B., WESTIN S., COBB J.E., LAMBERT M.H., KUROKAWA R., ROSENFELD M.G., WILLSON T.M., GLASS C.K., MILBURN M. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- O'BANION M.K., WINN V.D., YOUNG D.A. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4888–4892. doi: 10.1073/pnas.89.11.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OBERFIELD J.L., COLLINS J.L., HOLMES C.P., GOREHAM D.M., COOPER J.P., COBB J.E., LENHARD J.M., HULL-RYDE E.A., MOHR C.P., BLANCHARD S.G., PARKS D.J., MOORE L.B., LEHMANN J.M., PLUNKET K., MILLER A.B., MILBURN M.V., KLIEWER S.A., WILLSON T.M. A peroxisome proliferator-activated receptor γ ligand inhibits adipocyte differentiation. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6102–6106. doi: 10.1073/pnas.96.11.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REGINATO M.J., BAILEY S.T., KRAKOW S.L., MINAMI C., ISHII S., TANAKA H., LAZAR M.A. A potent antidiabetic thiazoldinedione with unique peroxisome proliferator-activated receptor γ- activating properties. J. Biol. Chem. 1998a;273:32679–32684. doi: 10.1074/jbc.273.49.32679. [DOI] [PubMed] [Google Scholar]
- REGINATO M.J., KRAKOW S.L., BAILEY S.T., LAZAR M.A. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J. Biol. Chem. 1998b;273:1855–1858. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- RICOTE M., HUANG J., FAJAS L., LI A., WELCH J., NAJIB J., WITZTUM J.L., AUWERX J., PALINSKI W., GLASS C.K. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidised low density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 1998b;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICOTE M., LI A.C., WILLSON T.M., KELLY C.J., GLASS C.K. The peroxisome proliferator-activated receptor γ is a negative regulator of macrophage activation. Nature. 1998a;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- RISTOW M., MULLER-WIELAND D., PFEIFFER A., KRONE W., KAHN C.R. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. New. Eng. J. Med. 1998;339:953–959. doi: 10.1056/NEJM199810013391403. [DOI] [PubMed] [Google Scholar]
- ROSS R. Atherosclerosis–an inflammatory disease. New Eng. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- RUSCHITZKA F.T., NOLL G., LUSCHER T.F. The endothelium in coronary artery disease. Cardiology. 1997;88:3–19. doi: 10.1159/000177500. [DOI] [PubMed] [Google Scholar]
- SARRAF P., MUELLER E., JONES D., KING F.J., DEANGELO D.J., PARTRIDGE J.B., HOLDEN S.A., CHEN L.B., SINGER S., FLETCHER C., SPIEGELMAN B.M. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nature Med. 1998;4:1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- SATOH H., TSUKAMOTO K., HASHIMOTO Y., HASHIMOTO N., TOGO M., HARA M., MAEKAWA H., ISOO N., KIMURA S., WATANABE T. Thiazoldinediones suppress endothelin-1 secretion from bovine endothelial cells: a new possible role for PPARγ on vascular endothelial function. Biochem. Biophys. Res. Commun. 1999;254:757–763. doi: 10.1006/bbrc.1998.0126. [DOI] [PubMed] [Google Scholar]
- SCHMIDT A., ENDO N., RUTLEDGE S.J., VOGEL R., SHINAR D., RODAN G.A. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 1992;6:1634–1641. doi: 10.1210/mend.6.10.1333051. [DOI] [PubMed] [Google Scholar]
- SHER T., YI H-F., MCBRIDE O.W., GONZALEZ F.J. cDNA cloning, chromosomal mapping, and functional characterisation of the human peroxisome proliferator-activated receptor. Biochemistry. 1993;32:5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- SIGAL E., LAUGHTON C.W., MULKINS M.A. Oxidation, lipoxygenase, and atherogenesis. Ann. N.Y. Acad. Sci. 1994;714:211–224. doi: 10.1111/j.1749-6632.1994.tb12046.x. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., DEWITT D.L., ALLEN M.L. Bimodal distribution of the PGI2 synthase antigen in smooth muscle cells. J. Biol. Chem. 1983;258:4922–4926. [PubMed] [Google Scholar]
- SMITH W.L., MARNETT L.J., DEWITT D.L. Prostaglandin and thromboxane biosynthesis. Phamac. Ther. 1991;49:153–179. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- SONG J., WALSH M.F., IGWE R., RAM J.L., BARAZI M., DOMINGUEZ L.J., SOWERS J.R. Troglitizone reduces contraction by inhibition of vascular smooth muscle cell Ca2+ currents and not endothelial nitric oxide production. Diabetes. 1997;46:659–664. doi: 10.2337/diab.46.4.659. [DOI] [PubMed] [Google Scholar]
- SPIEGELMAN B.M. PPARγ: Adipogenic regulator and thiazoldinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- SPIEGELMAN B.M., FLIER J.S. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- STAELS B., DALLONGEVILLE J., AUWERX J., SCHOONJANS K., LEITERSDORF E., FRUCHART J-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998b;98:2088–2093. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- STAELS B., KOENIG W., HABIB A., MERVAL R., LEBRET M., TORRA I.P., DELERIVE P., FABEL A., CHINETTI G., FRUCHART J-C., NAJIB J., MACLOUF J., TEDGUI A. Activation of human aortic smooth muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998a;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- STEINBERG D. Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- TONTONOZ P., NAGY L., ALVAREZ J.G.A., THOMAZY V.A., EVANS R.M. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidised LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- TORCHIA J., GLASS C., ROSENFELD M.G. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 1998;10:373–383. doi: 10.1016/s0955-0674(98)80014-8. [DOI] [PubMed] [Google Scholar]
- TSUJII M., KAWANO S., TSUJI S., SAWAOKA H., HORI M., DUBOIS R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- TUGWOOD J.D., ISSEMANN I., ANDERSON R.G., BUNDELL K.R., MCPHEAT W.L., GREEN S. The mouse peroxisome proliferator-activated receptor recognises a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992;11:433–439. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UPPENBERG J., SVENSSON C., JAKI M., BERTILSSON G., JENDEBERG L., BERKENSTAM A. Crystal structure of the ligand binding domain of the human nuclear receptor PPARγ. J. Biol. Chem. 1998;273:31108–31112. doi: 10.1074/jbc.273.47.31108. [DOI] [PubMed] [Google Scholar]
- XIN X., YANG S., KOWALSKI J., GERRITSEN M.E. Peroxisome proliferator-activated receptor γ ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999;274:9116–9121. doi: 10.1074/jbc.274.13.9116. [DOI] [PubMed] [Google Scholar]
- XU H.E., LAMBERT M.H., MONTANA V.G., PARKS D.J., BLANCHARD S.G., BROWN P.J., STERNBACH D.D., LEHMANN J., WISELY G.B., WILLSON T.M., KLIEWER S.A., MILBURN Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- YLA-HERTTUALA S., ROSENFELD M.E., PARTHASARATHY S., GLASS C.K., SIGAL E., WITZTUM J.L., STEINBERG D. Co-localisation of 15-lipoxygenase mRNA and protein with epitopes of oxidised LDL in macrophage-rich areas of atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6959–6963. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YLA-HERTTUALA S., ROSENFELD M.E., PARTHASARATHY S., SIGAL E., SARKIOJA T., WITZTUM J.L., STEINBERG D. Gene expression in macrophage-rich human atherosclerotic lesions. 15-lipoxygenase and acetyl low density lipoprotein receptor messenger RNA colocalize with oxidation specific- lipid protein aducts. J. Clin. Invest. 1991;87:1146–1152. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU G.S., LU Y.C., GULICK T. Co-regulation of tissue specific alternative human carnitine palmitoyltransferase Iβ gene promoters by fatty acid enzyme substrate. J. Biol. Chem. 1998;273:32901–32909. doi: 10.1074/jbc.273.49.32901. [DOI] [PubMed] [Google Scholar]
- YU K., BAYONA W., KALLEN C.B., HARDING H.P., RAVERA C.P., MCMAHON G., BROWN M., LAZAR M. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- ZHANG F., SOWERS R.J., RAM J.L., STANDLEY P.R., PEULER J.D. Effects of pioglitazone on calcium channels in vascular smooth muscle. Hypertension. 1994;24:170–175. doi: 10.1161/01.hyp.24.2.170. [DOI] [PubMed] [Google Scholar]
- ZHU Y., ALVARES K., HUANG Q., RAO M.S., REDDY J.K. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 1993;268:26817–26820. [PubMed] [Google Scholar]
- ZHU Y., QI C., CALANDRA C., RAO M.S., REDDY J.K. Cloning and identification of mouse steriod receptor co-activator-1 (mSRC-1), as a co-activator or peroxisome proliferator-activated receptor-γ. Gene Exp. 1996;6:185–195. [PMC free article] [PubMed] [Google Scholar]
- ZHU Y., Qi C., JAIN S., RAO M.S., REDDY J.K. Isolation and characterisation of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem. 1997;272:25500–25506. doi: 10.1074/jbc.272.41.25500. [DOI] [PubMed] [Google Scholar]