

Abstract
Anthracyclines can cause cumulative dose-related cardiotoxicity characterized by changes in Ca2+ metabolism, including dysfunction of the sacroplasmic reticulum (SR) and decreased expression of Ca2+-handling proteins, such as the ryanodine receptor (RyR2). In this study, we examined the effect of dexrazoxane (ICRF-187), an iron chelator which prevents anthracycline cardiotoxicity, on RyR2 gene expression in rats treated chronically with daunorubicin. Daunorubicin (2.5 mg kg−1 i.v. weekly for 6 weeks) produced cardiotoxicity as demonstrated by histopathologic changes. The ryanodine receptor/glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA ratio was decreased by 38±3% (P<0.02) compared to values in control rats. Dexrazoxane pre-treatment (50 mg kg−1; 1 h prior to each daunorubicin injection) prevented the decrease in RyR2/GAPDH mRNA ratio and histopathologic lesions in daunorubicin-treated rats. This is the first report that a protective agent such as dexrazoxane can ameliorate the decreased expression of a specific gene involved in anthracycline-induced cardiotoxicity.
Keywords: Ryanodine receptor, anthracycline cardiotoxicity, sarcoplasmic reticulum, dexrazoxane, cardioprotectant
Introduction
Anthracyclines are among the most important cancer chemotherapeutic agents, owing to their efficacies against acute leukaemias, lymphomas, breast cancer, lung cancer, and soft tissue sarcomas (Young et al., 1981). Chronic anthracycline therapy, however, results in impairment of myocardial function, but the cellular mechanisms remain enigmatic (Olson & Mushlin, 1990). Sacroplasmic reticulum dysfunction appears to play a significant role in anthracycline cardiomyopathy (Dodd et al., 1993; Phillips et al., 1998), since decreases in myocardial contractility are associated with impaired Ca2+ homeostasis and perturbations of the SR Ca2+ release channel (ryanodine receptor) (Dodd et al., 1993; Arai et al., 1998; Gambliel et al., 1999). Chronic anthracycline administration also causes decreased gene expression of the SR calcium release channel (Dodd et al., 1993; Arai et al., 1998). This down-regulation is an early event, occurring prior to perturbations of other SR calcium handling proteins (Gambliel et al., 1999). Thus, a decrease in ryanodine receptor mRNA levels may play a significant role in doxorubicin cardiotoxicity. This hypothesis would predict that prevention of anthracycline cardiotoxicity would be accompanied by prevention of down-regulation of RyR2 gene expression. Such an observation would further implicate the role of down-regulation of RyR2 in anthracycline cardiomyopathy. To address this question, we designed studies to determine whether pre-treatment with dexrazoxane, an established protectant against anthracycline cardiotoxicity (Villani et al., 1990; Yeung et al., 1992; Herman et al., 1993) prevents reduction of gene expression of RyR2 in rats following chronic daunorubicin treatment.
We report that dexrazoxane pre-treatment prevented down-regulation of the cardiac SR calcium release channel (RyR2) mRNA levels normalized to glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA levels in rats chronically treated with daunorubicin. Dexrazoxane pre-treatment also ameliorated the histopathologic lesions in left ventricular myocardium caused by chronic daunorubicin administration. Thus, an intervention which reduces anthracycline cardiotoxicity also opposes down-regulation of gene expression of RyR2. This observation supports the idea that down-regulation of RyR2 may be involved in the pathogenesis of anthracycline cardiotoxicity.
Methods
Adult (4–6 months) male Fisher 344 rats were randomly allocated into one control and three treatment groups. Rats treated with daunorubicin received sodium lactate (dexrazoxane vehicle; 18.7 mg kg−1), 1 h prior to daunorubicin injection (2.5 mg kg−1, i.v. bolus, once a week for 6 weeks). Rats treated with daunorubicin and dexrazoxane received dexrazoxane (50 mg kg−1, i.v.) 1 h prior to daunorubicin injections as in the previous group. The third treatment group received the daunorubicin vehicle (0.9% saline) 1 h after the dexrazoxane injection (50 mg kg−1, i.v., once a week for 6 weeks). Rats in the vehicle control group received the dexrazoxane and daunorubicin vehicles. The mean daily food intake of rats in the other three groups was matched to the quantity of food consumed by the daunorubicin-treated group. Two weeks after the last injection, rats were sacrificed by decapitation and their hearts immediately excised. Apical tissue samples were preserved in 10% neutral buffered formalin and used to determine histopathologic scores by a pathologist blinded to treatment. Alterations in histology were graded on a scale of 0 to 4, based on the percentage of cells showing vacuolization and myofibrillar loss (Herman & Ferrans, 1993). If there was no damage, a score of 0 was assigned; less than 10%, 1; 10–24%, 2; 25–49%, 3; and if greater than 50% of cells showed damage, a score of 4 was assigned. A minimum of two sections from each heart and five fields per section were quantitated per rat.
The rat cardiac SR calcium release channel (RyR2) and glyceraldehyde phosphate dehydrogenase (GAPDH) partial cDNAs were obtained from total left ventricular RNA by RT–PCR using the following primer pairs: RyR2; 5′-cgcgaattcgtcttccttgacttggtcttg-3′ and 5′-cgcgaattcagggaggatggtgacacgccg-3′; GAPDH; 5′-cgcgaattcttactccttggaggcatgta-3′ and 5′-cgcgaattcatggtgaaggtcggtgtcaac-3′. The PCR products for RyR2 (247 bp, codons 41–288) and GAPDH (988 bp, codons 35–1018) were subcloned in pCR2.1 (Invitrogen). Immediately after removing the heart, 50–100 mg of left ventricular free wall was frozen in liquid nitrogen until needed for preparation of total RNA. The tissue was homogenized in Chaosolv reagent (Ultraspec RNA reagent, Biotex, Houston, TX, U.S.A.) using a Brinkman ploytron homogenizer (3×30 s, setting 7) (Chomczyinski & Sacchi, 1987). Total RNA was precipitated in ethanol and stored at −80°C until needed for assay. Prior to RNase protection analysis, RNA was quantitated spectrophotometrically at 260 nm. The integrity of the 18 s and 28 s RNA bands, and RNA loads were also verified by agarose electrophoresis. The plasmids encoding the RyR2 and GAPDH probes were linearized with BGl II and Dde 1 (Stratagene) respectively, and cRNA 32P-uridinyl triphosphate radiolabelled probes synthesized using the T7 promoter with an in vitro transcription reagent kit (Stratagene, La Jolla, CA, U.S.A.). After synthesis, the probes were treated with 1 unit RQ1 DNAse, extracted with phenol and chloroform, ethanol precipitated, electrophoresed, and eluted from a non-denaturing 6% polyacrylamide gel according to standard procedure (Gilman, 1987).
RNase protection assays (RPAs) were performed using kit reagents from Boehringer Mannheim (Indianapolis, IN, U.S.A.). 32P-cRNAs (200,000 c.p.m. per probe) were hybridized for 16 h at 45°C with 4 μg total ventricular RNA. RNase digestion conditions were as recommended by the manufacturer. Protected RNA fragments were ethanol precipitated, denatured, and resolved on a 6% polyacrylamide gel containing 8 M urea. After drying, the gel was subjected to autoradiography and developed utilizing varying exposures. Protected cRNA fragments (134-nt, RyR2; 183-nt, GAPDH) were quantitated by densitometry after evaluation of exposures to obtain an adequate dynamic range of linear response. Densitometric scanning of autoradiograms was performed using Imagequant software on a Molecular Dynamics (Sunnyvale, CA, U.S.A.) scanning instrument.
Animal care and use in this study were performed in accordance with NIH guidelines. Densitometric results were expressed as the ratio of RyR2 to GAPDH mRNA for each rat. Experiments were performed in duplicate. Comparisons of mean values were made using one-way ANOVA and Tukey's Test for All Pairwise Multiple Comparisons (SigmaStat, SPSS, Inc.), with alpha set at 0.05.
Results
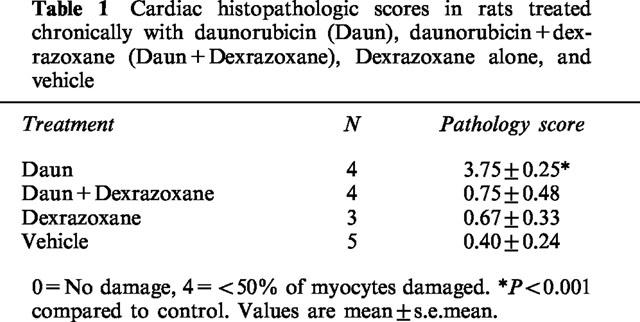
Figure 1A demonstrated the tissue specificity of the RyR2 probe. GAPDH mRNA was present in approximately equal amount in liver and cardiac tissue from untreated rats. RyR2 mRNA was specific to cardiac tissue; no observable amounts were detected in liver. Figure 1B is a representative composite autoradiograph made from a single gel of RPAs of GAPDH and RyR2 mRNA obtained from cardiac tissue. Daunorubicin treatment alone caused a reduction in RyR2 mRNA. Quanitation by densitometric scanning (Figure 1C) demonstrated that myocardial RyR2/GAPDH mRNA ratios in daunorubicin-treated animals were decreased by 38±3% (mean±s.e.mean) compared with vehicle-treated control values (P<0.02). Dexrazoxane pre-treatment prevented the daunorubicin-induced decrease in RyR2 gene expression. Dexrazoxane treatment alone had no effect on RyR2/GAPDH mRNA ratios. Histopathologic scoring demonstrated a similar effect by dexrazoxane (Table 1). Histologic lesions in daunorubicin-treated animals included vacuolization, myofibrillar necrosis, and perivascular fibrosis. Dexrazoxane prevented (P<0.01) the histopathologic damage observed in daunorubicin-treated animals.
Figure 1.
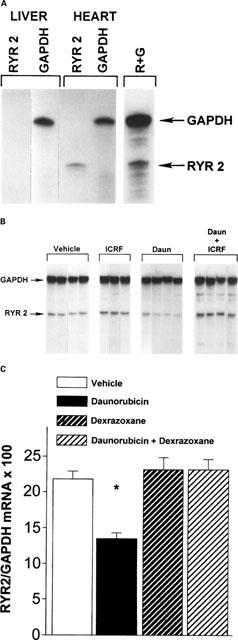
Protective effects of dexrazoxane pre-treatment on daunorubicin-induced decreases in RyR2 gene expression in hearts from chronically treated rats. Abbreviations are: daunorubicin treated, dexrazoxane vehicle pre-treated rats, Daun (n=4); dexrazoxane pre-treated, daunorubicin treated rats, Daun+ICRF (n=4); dexrazoxane pre-treated, daunorubicin vehicle treated rats, ICRF (n=3); daunorubicin and dexrazoxane vehicle treated rats, Vehicle (n=5). (A) shows that the RyR2 probe is specific for the cardiac isoform only; none was detected in the liver fractions. The probe for GAPDH binds equally well to mRNA from liver or heart. Lane 5 shows both probes in the same hybridization reaction with cardiac mRNA. (B) is a representative composite autoradiograph made from a single gel of a RPA of cardiac tissue GAPDH and RyR2 mRNA. Daunorubicin treatment produced decreases in RyR2 mRNA (lanes 8–11). Dexrazoxane pre-treatment prevented daunorubicin-induced reductions in RyR2 mRNA (lanes 12–15). There was no change in RyR2 mRNA with dexrazoxane treatment alone (lanes 5–7). GAPDH was not significantly altered by treatment. (C) is a histogram of data obtained from densitometric scans of autoradiographs from the RPA analysis. Pre-treatment with dexrazoxane prevented the daunorubicin-induced decrease in RyR2/GAPDH mRNA ratio. Dexrazoxane alone had no effect on the RyR2-GAPDH mRNA ratio. Values are mean±s.e.mean, for per cent RyR2/GAPDH mRNA expressed in arbitrary units. Experiments were run in duplicate. *Different from control values; P<0.02.
Table 1.
Cardiac histopathologic scores in rats treated chronically with daunorubicin (Daun), daunorubicin+dexrazoxane (Daun+Dexrazoxane), Dexrazoxane alone, and vehicle
Discussion
Chronic daunorubicin administration to rats produced cardiotoxicity as demonstrated by marked changes in histopathology, including myofibrillar loss, vacuolization, and perivascular fibrosis, as well as reduced RyR2 gene expression in ventricular tissue. Dexrazoxane pre-treatment prevented the histologic damage and the decrease in ryanodine gene receptor induced by daunorubicin. This finding further supports the role of altered ryanodine receptor gene expression in anthracycline cardiomyopathy (Dodd et al., 1993; Arai et al., 1998; Gambliel et al., 1999).
Dexrazoxane is hydrolyzed intracellularly into a bidendate iron-chelator resembling EDTA (Yeung et al., 1992). It is hypothesized to exert its cardioprotective action in anthracycline cardiomyopathy through iron chelation (Minotti et al., 1998). The interaction of dexrazoxane with iron, and our current finding of dexrazoxane's effect on an important gene regulating calcium metabolism suggest a possible interaction between iron metabolism and gene expression of SR calcium handling proteins in anthracycline-induced cardiomyopathy. Low molecular weight iron complexes have been shown to be cardiotoxic (Anghileri et al., 1995) and to directly bind and inhibit calcium release from SR (Kim et al., 1995). The present study suggests that iron also may affect calcium metabolism by regulating expression of the SR calcium release channel.
The C-13 hydroxy metabolite of doxorubicin, doxorubicinol, has been shown to delocalize Fe (II) from the iron-sulphur cluster of cytoplasmic aconitase, and to irreversibly inhibit aconitase and iron regulatory protein 1 (IRP-1) (Minotti et al., 1999). Normally, iron-sulphur cluster assembly-reassembly of aconitase is reversible and serves as a switch to modify transferrin receptor and ferrin mRNA expression, and regulate intracellular iron homeostasis. Thus, intramyocardial formation of doxorubicinol may contribute to oxidant stress through an iron-dependent mechanism. Several transcription factors have been observed to be affected by intracellular oxidant stress and iron (Kamata & Hirata, 1999), and such a mechanism may affect gene expression of calcium handling proteins. Additional studies are required to test the idea that daunorubicin administration disrupts iron regulation to decrease gene expression of RyR2. Nevertheless, this is the first report of a protective effect by the iron chelator, dexrazoxane, on expression of a specific gene considered to be important to the pathogenesis of anthracycline-induced cardiac failure.
Acknowledgments
This work was supported by Merit Review Grants of the Veterans Administration to R.D. Olson and B.J. Cusack. Funding was also provided by the MSTI/MSMRI Research Institute. We would like to thank Barbara Trajkovska, Mary Hicks, and Linda Zhang for their excellent technical assistance.
Abbreviations
- Ca2+
calcium
- EDTA
ethylenediaminetetraacetic acid
- GAPDH
glyceraldehyde phosphate dehydrogenase
- mRNA
messenger ribonucleic acid
- RPA
ribonuclease protection assay
- RT–PCR
reverse transcriptase-polymerase chain reaction
- RyR2
ryanodine receptor
- SR
sacroplasmic reticulum
References
- ANGHILERI L.J., MAINCENT P., THOUVENOT P. Cardiotoxicity of parenterally administered iron complexes. Arzneimittelforschung. 1995;45:679–681. [PubMed] [Google Scholar]
- ARAI M., TOMARU K., TAKIZAWA T., SEKIGUCHI K., YOKOYAMA Y., SUZUKI T., NAGAI R. Sacroplasmic reticulum genes are selectively down-regulated in cardiomyopathy produced by doxorubicin in rabbits. J. Mol. Cell. Cardiol. 1998;30:243–254. doi: 10.1006/jmcc.1997.0588. [DOI] [PubMed] [Google Scholar]
- CHOMCZINSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate phenol-chloroform extraction. Anal. Biochem. 1987;162:156–161. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- DODD D.A., ATKINSON J.B., OLSON R.D., BUCK S., CUSACK B.J., FLEISCHER S., BOUCEK R.J. Doxorubicin cardiomyopathy is associated with decrease in calcium release channel of SR in a chronic rabbit model. J. Clin. Invest. 1993;91:1697–1705. doi: 10.1172/JCI116379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAMBLIEL H., CUSACK B.J., TRAJKOVSKA B., MUDUMBI M., OLSON R.D. Dexrazoxane prevents systolic dysfunction in chronic daunorubicin cardiotoxicity in the Fischer 344 rat. Pharmacologist. 1999;41:120. [Google Scholar]
- GILMAN M.Ribonuclease Protection Assay Current Protocols in Molecular Biology 1987New York: John Wiley & Sons; 471–499.ed. Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Siedman, J.G., Smith, J.A. & Struhl, K. pp [Google Scholar]
- HERMAN E.H., FERRANS V.J. Timing of treatment of ICRF-187 and its effect on chronic doxorubicin cardiotoxicity. Cancer Chemother. Pharmacol. 1993;32:445–449. doi: 10.1007/BF00685888. [DOI] [PubMed] [Google Scholar]
- KAMATA H., HIRATA H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- KIM E., GIRI S.N., PESSAH I.N. Iron (II) is a modulator of ryanodine-sensitive calcium channels of cardiac muscle sacroplasmic reticulum. Toxicol. Appl. Pharmacol. 1995;130:57–66. doi: 10.1006/taap.1995.1008. [DOI] [PubMed] [Google Scholar]
- MINOTTI G., CAIRO G., MONTI E. Role of iron in anthracycline cardiotoxicity: new tune for an old song. FASEB J. 1999;13:199–212. [PubMed] [Google Scholar]
- MINOTTI G., RECALCATI S., MORDENTE A., LIBERI G., CALAFIORE A.M., MANCUSO C., PREZIOSI P., CAIRO G. The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cystolic fractions from human myocardium. FASEB J. 1998;12:541–552. doi: 10.1096/fasebj.12.7.541. [DOI] [PubMed] [Google Scholar]
- OLSON R.D., MUSHLIN P.S. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB J. 1990;4:3076–3086. [PubMed] [Google Scholar]
- PHILLIPS R.M., NARAYAN P., GOMEZ A.M., DILLY K., JONES L.R., LEDERER W.J., ALTSCHULD R.A. Sarcoplasmic reticulum in heart failure: central player or bystander. Cardiovascular Res. 1998;37:346–351. doi: 10.1016/s0008-6363(97)00260-5. [DOI] [PubMed] [Google Scholar]
- VILLANI F., GALINBERLI M., MANH E., COVA D., LANZA E., ROZZA-DIONIGI A., FAVALLI L., POGGI P. Effect of ICRF-187 pretreatment against doxorubicin-induced delayed cardiotoxicity in the rat. Toxicol. Appl. Pharmacol. 1990;102:292–299. doi: 10.1016/0041-008x(90)90028-s. [DOI] [PubMed] [Google Scholar]
- YEUNG T.K., JAENKE R.S., WILDING D., CREIGHTON A.M., HOPEWELL J.W. The protective activity of ICRF-187 against doxorubicin-induced cardiotoxicity in the rat. Cancer Chemother. Pharmacol. 1992;30:58–64. doi: 10.1007/BF00686486. [DOI] [PubMed] [Google Scholar]
- YOUNG R.C., OZOLS R.F., MYERS C.E. The anthracycline antineoplastic drugs. N. Engl. J. Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]