

Abstract
Endotoxin is known to confer a delayed protection against myocardial infarction. Lipopolysaccharide (LPS) treatment also induces the de novo synthesis of kinin B1-receptors that are not present in normal conditions. The aim of this study was to evaluate whether LPS-induced B1-receptors are implicated in the reduction of infarct size brought about by LPS.
Rabbits were submitted to a 30-min coronary artery occlusion and 3-h reperfusion sequence. Six groups were studied: pretreated or not (control animals) with LPS (5 μg kg−1 i.v.) 24 h earlier and treated 15 min before and throughout ischaemia–reperfusion with either the B1-antagonist R-715 (1 mg kg−1 h−1), the B1-agonist Sar-[D-Phe8]-des-Arg9-bradykinin (15 μg kg−1 h−1) or vehicle (saline). Infarct size and area at risk were assessed by differential staining and planimetric analysis.
The presence of B1-receptors in LPS-pretreated animals was confirmed by a decrease in mean arterial pressure in response to B1 stimulation. LPS-pretreatment significantly reduced infarct size (6.4±1.7%, of area at risk vs 24.1±2.5% in control animals, P<0.05). This protection was not modified by B1-receptor antagonism (7.4±2.2%, NS) or stimulation (5.2±1.2%, NS). Neither antagonist nor agonist modified infarct size in control animals.
In conclusion, these data suggest that LPS-induced myocardial protection in the rabbit is not related to concomitant de novo B1-receptor induction.
Keywords: Kinin B1-receptors, endotoxin, myocardial protection
Introduction
Endotoxin is known to depress myocardial function but can also induce a delayed myocardial protection against subsequent endotoxic shock (Meng et al., 1996) or infarction (Brown et al., 1989). In the rabbit, 72 h after bacterial lipopolysaccharide (LPS) treatment, the infarct size induced by a myocardial ischaemia–reperfusion sequence is reduced (Rowland et al., 1996). In the rat, pretreatment with LPS decreases the ischaemic insult thus improving cardiac function (Rowland et al., 1997) and reducing cell injury (Zacharowski et al., 1999), infarct size (Eising et al., 1996) and arrhythmia incidence (Song et al., 1994). This protection is related to protein synthesis since it is inhibited by cycloheximide (Meng et al., 1997). Various protective mechanisms have been proposed such as an increase in antioxidant enzyme activity (Brown et al., 1989; Maulik et al., 1995), synthesis of heat stress proteins (Rowland et al., 1996; Meng et al., 1996) and induction of nitric oxide synthase (McKenna et al., 1995).
B1-receptors are not present in normal conditions but are induced by various inflammatory stimuli such as in vitro incubation (Regoli et al., 1977) and in vivo administration of LPS (Regoli et al., 1981) or cytokines (deBlois et al., 1989). Vascular B1-receptors are present in the rabbit 5 and 20 h after LPS administration (Regoli et al., 1981). Their induction is prevented by pretreatment with protein synthesis inhibitors (deBlois et al., 1989). When stimulated, B1-receptors induce a hypotension due to an endothelial-dependent vasodilation (Regoli et al., 1981; Pruneau & Bélichard, 1993). This could lead to improved cardiac function during myocardial ischaemia–reperfusion, especially by counteracting the no-reflow phenomenon. Another cardioprotective mechanism associated with B1-receptor stimulation could be brought about by their ability to reduce noradrenaline release upon ischaemia–reperfusion (Chahine et al., 1993; Feng et al., 1997).
Kinins, which are generated during myocardial ischaemia–reperfusion (Pitt et al., 1970; Torstila, 1978), are known to participate in ischaemic preconditioning mostly by stimulating B2-receptors (Brew et al., 1995; Parratt et al., 1995). However B1-receptors have been implicated in the early protective effects on endothelial function (Bouchard et al., 1998).
The aim of this study was thus to investigate whether B1-receptors, induced by LPS, could participate to the delayed myocardial protection conferred by this toxin. To evaluate this, we tested the effect of B1-receptor blockade and stimulation on the development of infarct size in LPS-pretreated rabbits.
Methods
Surgical preparation
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication No. 85-23, revised 1985).
Specific pathogen-free male New-Zealand rabbits (ESD, France, n=42) weighing 2.6–3.8 kg, were anaesthetized with pentobarbitone sodium (40 mg kg−1) administered via the marginal ear vein. Additional doses were given as required. Positive pressure respiration with room air was maintained by a pump (Roche-Kontron 3100S) connected to an endotracheal tube. Ventilation rate and tidal volume were respectively 35 breaths min−1 and 30 ml.
The right common carotid artery was cannulated with a polyethylene catheter and the arterial pressure was measured using a pressure transducer (Baxter 33-260, Healthcare Corp., U.K.). The right jugular vein was cannulated for intravenous drug administration. A left thoracotomy was performed at the 4th intercostal space and the pericardium was opened. A 3/0 silk thread was then placed around the first marginal branch of the circumflex artery and passed through a small polyethylene tube.
Heart rate and arterial pressure were continuously recorded on a computer using a data acquisition system (Power Lab, ADInstruments). An intravenous injection of 1000 I.U. of heparin (Choay) was performed at the beginning of the experimental protocol.
Experimental protocol
After the surgical preparation, a 15 min stabilization period was observed. The animals then received the various drug treatments starting 15 min before and throughout a 30-min occlusion and 3-h reperfusion period. Clamping the snare around the artery produced ischaemia; successful ligation was confirmed by myocardial cyanosis with bulging and reperfusion by the appearance of a myocardial blush (Marber et al., 1993).
Area at risk and infarct size measurement
At the end of the protocol, the heart was removed for infarct size assessment using a variant of the classic method described by Marber et al. (1993). The heart was retrogradly perfused via the aorta with a physiological solution adjusted to pH 7.4 containing (in mM): NaCl 118, NaHCO3 25, KCl 4.7, MgSO4 1.22, KH2PO4 1.2, CaCl2 1.8, glucose 11. The coronary artery was re-occluded and a blue dye (Unisperse blue, Ciba Geigy) was perfused in the non-ischaemic zone. The left ventricle was frozen and cut in 2 mm slicesthat were incubated for 10 min at 37°C in phosphate buffer containing 1% triphenyl tetrazolium chloride (Sigma). The area at risk and infarct size were measured using a computerized planimetric technique (Minichromax, Biolab) and expressed as a percentage of the left ventricle and area at risk, respectively.
Peptide synthesis
Sar-[D-Phe8]-des-Arg9-bradykinin and AcLys-[D-βNal7, Ile8]-des-Arg9-bradykinin (R-715) were synthesized with a peptide synthesizer (Applied Biosystems 430 A) using Merrifield-type resins with the first amino acid attached. Amino acids were activated by dicyclohexylcarbodiimide 1-hydroxybenzotriazole (Peptides International) on 1-methyl-2-pyrrolidinone. Peptides were cleaved from the resins with anhydrous hydrogen fluoride in the presence of appropriate scavengers. The resulting peptides were purified by medium-performance reversed-phase (C18) chromatography and if necessary by HPLC. Peptide purity and identity were confirmed by analytical HPLC and by ion-mass spectrometry respectively, as described by Drapeau & Regoli (1988).
Experimental groups
Endotoxin treatment, at a dose (5 μg kg−1) known to induce B1-receptors in the rabbit (Regoli et al., 1981), was produced by injecting LPS (from E. coli serotype 0111:B4, Sigma) via the marginal ear vein 24 h before the experimental protocol. Six groups of animals (n=7 in each) were studied in which control and LPS-pretreated animals were submitted to one of the following treatments administered intravenously at a rate of 5 ml h−1 15 min before and throughout the occlusion–reperfusion period: (1) Saline perfusion; (2) Perfusion with the B1-receptor antagonist R-715 at a dose of 1 mg kg−1 h−1 in saline; or (3) Perfusion with the B1-receptor agonist Sar-[D-Phe8]-des-Arg9-bradykinin at a dose of 15 μg kg−1 h−1 in saline.
Statistical analysis of data
In order to compare data from experimental groups, three- and two-way analyses of variance followed by post hoc multiple comparison Tukey tests were performed when normality was respected. When it was not, non-parametric Kruskal-Wallis tests with multiple comparison Dunn's tests and Mann-Whitney Rank Sum tests were used for intra and inter-group analyses respectively (SigmaStat statistical software v.2.0, Jandel Scientifics). Statistical significance was set at P⩽0.05.
Results
Haemodynamic measurements
All animals survived the LPS treatment and the ischaemia–reperfusion protocol. They were therefore all included in the study.
Pretreatment with LPS resulted in significantly decreased arterial pressure values before and during ischaemia but not upon reperfusion (Table 1). In LPS-pretreated animals, treatment with the B1-agonist Sar-[D-Phe8]-des-Arg9-bradykinin significantly decreased both mean arterial pressure and heart rate in comparison to control animals (Figure 1A,B). Treatment with the B1-antagonist R-715 slightly but significantly increased mean arterial pressure in LPS-pretreated animals (Figure 1A). This was however not sufficient to bring the mean arterial pressure back to the control value. Indeed, neither B1-agonist nor antagonist significantly modified the arterial pressure response to the ischaemia-reperfusion protocol.
Table 1.
Variations of mean arterial pressure before during and after coronary occlusion
Figure 1.
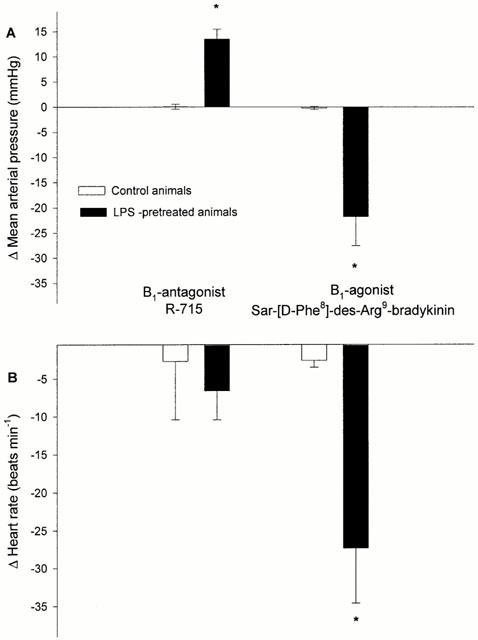
Decreased mean arterial pressure (A) and heart rate (B) (mean±s.e.mean) 15 min after the beginning of Sar-[D-Phe8]-des-Arg9-bradykinin administration in lipopolysaccharide (LPS)-pretreated animals (n=7) and increased mean arterial pressure 15 min after R-715 administration. *P<0.05 vs corresponding control animals (n=7).
Infarct size
There was no significant difference in the area at risk (expressed as a percentage of the left ventricle) of the various experimental groups (Figure 2A).
Figure 2.
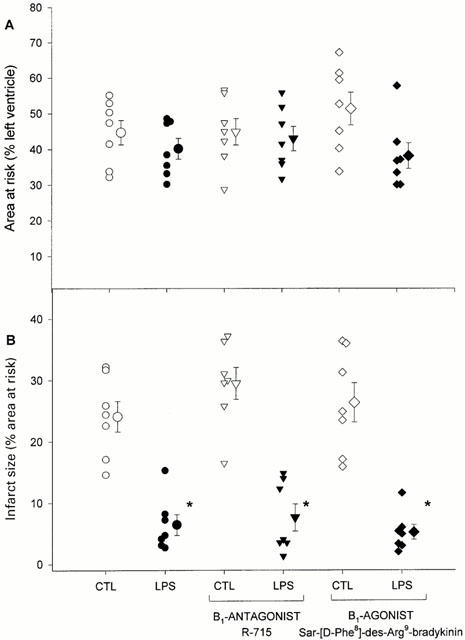
(A) Area at risk (expressed as a percentage of the left ventricle) was identical in all experimental groups. (B) Infarct size (expressed as a percentage of the area at risk) was reduced in the LPS-pretreated animals, independently of B1-receptor blockade or stimulation. Data are individual values and means±s.e.mean of CTL=control animals, LPS=lipopolysaccharide-pretreated animals. *P<0.05 vs corresponding CTL value.
LPS-pretreatment brought about an important (about 75%) reduction in infarct size (Figure 2B). The administration of B1-antagonist or agonist did not modify infarct size in either control or LPS-pretreated animals (Figure 2B).
Discussion
The present study demonstrates that kinin B1-receptors are not implicated in the endotoxin-induced delayed protection against myocardial infarction in the rabbit.
In our experimental conditions, LPS treatment dramatically decreased infarct size in all groups studied. This is in accordance with other studies in the rat (Zacharowski et al., 1999) or in the rabbit (Rowland et al., 1996). LPS treatment has been shown to induce synthesis of various cell protective proteins such as heat shock proteins (Meng et al., 1996), antioxidant enzymes like superoxide dismutase, catalase and glutathione reductase (Maulik et al., 1995), inducible nitric oxide synthase (McKenna et al., 1995) or cyclo-oxygenase 2 (Breder & Saper, 1996) that could participate in its beneficial effects against myocardial ischaemia–reperfusion injury.
The LPS-pretreatment carried out in this study was effective in inducing B1-receptors since, in accordance with previous studies, the administration of the B1-agonist Sar-[D-Phe8]-des-Arg9-bradykinin decreased arterial pressure and heart rate in pretreated animals (Audet et al., 1997). The absence of B1-receptors in control animals was confirmed by the lack of response to Sar-[D-Phe8]-des-Arg9-bradykinin or to R-715, both highly selective for this receptor (Gobeil et al., 1996; Audet et al., 1997). The hypertensive response seen in LPS-pretreated animals following B1-receptor blockade with R-715 suggests a basal stimulation of B1-receptors by endogenous agonists such as des-Arg9 derivatives. However, treatment with R-715 was not sufficient to bring the arterial pressure back to its control value. The hypotension observed after endotoxin treatment could rather be due to the vasodilatation associated with induction of nitric oxide synthase (Forfia et al., 1998).
We have shown that LPS was able to simultaneously, (1) induce B1-receptor synthesis and (2) afford a protection against myocardial ischaemia, 24 h after its administration. However, blockade or stimulation of B1-receptors throughout the ischaemia–reperfusion sequence did not modify infarct size in both LPS-pretreated and control animals. Thus B1-receptors do not appear to influence the development of cell injury during myocardial ischaemia-reperfusion.
Monophosphoryl lipid A (MLA) is a derivative of LPS presenting the same protective effects against myocardial ischaemia–reperfusion without other side effects (Elliott, 1998). We have recently demonstrated that MLA fails to induce B1-receptors 24 h after administration (time at which it is effective for myocardial protection) (Mazenot et al., 1999). This is another observation showing that induction of B1-receptors does not appear to be necessary for this type of cardioprotection. Moreover, a common property of LPS and MLA that could be responsible for their cardioprotective effects is their ability to induce nitric oxide synthase (McKenna et al., 1995; Zhao et al. 1997). Indeed, inducible nitric oxide synthase appears to be essential for MLA cardioprotection (Xi et al., 1999). Finally, the hypotension observed 24 h after LPS pretreatment could, by itself, have cardioprotective actions.
Although B1-receptors do not appear to affect the development of infarct size, they could present other protective effects in the setting of myocardial ischaemia–reperfusion. Indeed, they have been shown to decrease ischaemia-induced noradrenaline release in the rat (Chahine et al., 1993; Feng et al., 1997) but not in other species (Hatta et al., 1999). This discrepancy could be due to the lack of specific induction of B1-receptors in both cases. Further studies are thus needed to evaluate the exact role of B1-receptors in the context of myocardial ischaemia–reperfusion with special emphasis on their specific effects on noradrenaline release or inflammation.
In conclusion, this study provides evidence that B1-receptors are not involved in LPS-induced protection against myocardial ischaemia–reperfusion injury in the rabbit.
Abbreviations
- CTL
control
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- MLA
monophosphoryl lipid A
References
- AUDET R., RIOUX F., DRAPEAU G., MARCEAU F. Cardiovascular effects of Sar-[D-Phe8]des-Arg9-bradykinin, a metabolically protected agonist of B1 receptor for kinins, in the anaesthetized rabbit pretreated with a sublethal dose of bacterial lipopolysaccharide. J. Pharmacol. Exp. Ther. 1997;280:6–15. [PubMed] [Google Scholar]
- BREDER C.D., SAPER C.B. Expression of inducible cyclooxygenase mRNA in the mouse brain after systemic administration of bacterial lipopolysaccharide. Brain Res. 1996;713:64–69. doi: 10.1016/0006-8993(95)01474-8. [DOI] [PubMed] [Google Scholar]
- BREW E.C., MITCHELL M.B., REHRING T.F., GAMBONI-ROBERTSON F., MCINTYRE R.C., HARKEN A.H., BANERJEE A. Role of bradykinin in cardiac functional protection after global ischemia–reperfusion in rat heart. Am. J. Physiol. 1995;269:H1370–H1378. doi: 10.1152/ajpheart.1995.269.4.H1370. [DOI] [PubMed] [Google Scholar]
- BOUCHARD J.F., CHOUINARD J., LAMONTAGNE D. Role of kinins in the endothelial protective effect of ischaemic preconditioning. Br. J. Pharmacol. 1998;123:413–420. doi: 10.1038/sj.bjp.0701619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN J.M., GROSSO M.A., TERADA L.S., WHITMAN G.J., BANERJEE A., WHITE C.W., HARKEN A.H., REPINE J.E. Endotoxin pretreatment increases endogenous myocardial catalase activity and decreases ischemia–reperfusion injury of isolated rat hearts. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2516–2520. doi: 10.1073/pnas.86.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAHINE R., ADAM A., YAMAGUCHI N., GASPO R., REGOLI D., NADEAU R. Protective effects of bradykinin on the ischaemic heart: implication of the B1 receptor. Br. J. Pharmacol. 1993;108:318–322. doi: 10.1111/j.1476-5381.1993.tb12802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEBLOIS D., BOUTHILLIER J., MARCEAU F. Pharmacological modulation of the up-regulated responses to des-Arg9-bradykinin in vivo and in vitro. Immunopharmacology. 1989;17:187–198. doi: 10.1016/0162-3109(89)90047-7. [DOI] [PubMed] [Google Scholar]
- DRAPEAU G., REGOLI D. Synthesis of bradykinin analogs. Methods Enzymol. 1988;163:263–272. doi: 10.1016/0076-6879(88)63025-4. [DOI] [PubMed] [Google Scholar]
- EISING G.P., MAO L., SCHMID-SCHÖNBEIN G.W., ENGLER R.L., ROSS J. Effects of induced tolerance to bacterial lipopolysaccharide on myocardial infarct size in rats. Cardiovasc. Res. 1996;31:73–81. [PubMed] [Google Scholar]
- ELLIOTT G.T. Monophosphoryl Lipid A-induced delayed preconditioning against cardiac ischaemia–reperfusion injury. J. Mol. Cell. Cardiol. 1998;30:3–17. doi: 10.1006/jmcc.1997.0586. [DOI] [PubMed] [Google Scholar]
- FENG J., YAMAGUCHI N., FOUCART S., CHAHINE R., LAMONTAGNE D., NADEAU R. Transient ischaemia inhibits nonexocytotic release of norepinephrine following sustained ischaemia in rat heart: is bradykinin involved. Can. J. Physiol. Pharmacol. 1997;75:665–670. doi: 10.1139/cjpp-75-6-665. [DOI] [PubMed] [Google Scholar]
- FORFIA P.R., ZHANG X., OCHOA F., XU X., BERNSTEIN R., SEHGAL P.B., FERRERI N.R., HINTZE T.H. Relationship between plasma NOx and cardiac and vascular dysfunction after LPS injection in anaesthetized dog. Am. J. Physiol. 1998;274:H193–H201. doi: 10.1152/ajpheart.1998.274.1.H193. [DOI] [PubMed] [Google Scholar]
- GOBEIL F., NEUGEBAUER W., FILTEAU C., JUKIC D., ALLOGHO S.N., PHENG L.H., NGUYEN-LE X.K., BLOUIN D., REGOLI D. Structure-activity studies of B1 receptor-related peptides. Antagonists. Hypertension. 1996;28:833–839. doi: 10.1161/01.hyp.28.5.833. [DOI] [PubMed] [Google Scholar]
- HATTA E., MARUYAMA R., MARSHALL S.J., IMAMURA M., LEVI R. Bradykinin promotes ischemic norepinephrine release in guinea pig and human hearts. J. Pharmacol. Exp. Ther. 1999;288:919–927. [PubMed] [Google Scholar]
- MARBER M.S., LATCHMAN D.S., WALKER J.M., YELLON D.M. Cardiac stress protein elevation 24 hours after brief ischaemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- MAULIK N., WATANABE M., ENGELMAN D., ENGELMAN R.M., KAGAN V.E., KISIN E., TYURIN V., CORDIS G.A., DAS D.K. Myocardial adaptation to ischemia by oxidative stress induced by endotoxin. Am. J. Physiol. 1995;269:C907–C916. doi: 10.1152/ajpcell.1995.269.4.C907. [DOI] [PubMed] [Google Scholar]
- MAZENOT C., RIBUOT C., DEMENGE P., GODIN-RIBUOT D. Monophosphoryl lipid A, a derivative of bacterial lipopolysaccharide, fails to induce B1-receptor-dependent responses to (des-Arg9)-bradykinin in the rabbit in vivo. Immunopharmacology. 1999;41:165–168. doi: 10.1016/s0162-3109(98)00065-4. [DOI] [PubMed] [Google Scholar]
- MCKENNA T.M., LI S., TAO S. PKC mediates LPS- and phorbol-induced cardiac cell nitric oxide synthase activity and hypocontractility. Am. J. Physiol. 1995;269:H1891–H1898. doi: 10.1152/ajpheart.1995.269.6.H1891. [DOI] [PubMed] [Google Scholar]
- MENG X., AO L., BROWN J.M., MELDRUM D.R., SHERIDAN B.C., CAIN B.S., BANERJEE A., HARKEN A.H. LPS induces late cardiac functional protection against ischemia independent of cardiac and circulating TNF-alpha. Am. J. Physiol. 1997;273:H1894–H1902. doi: 10.1152/ajpheart.1997.273.4.H1894. [DOI] [PubMed] [Google Scholar]
- MENG X., BROWN J.M., AO L., NORDEEN S.K., FRANKLIN W., HARKEN A.H., BANERJEE A. Endotoxin induces cardiac HSP70 and resistance to endotoxemic myocardial depression in rats. Am. J. Physiol. 1996;271:C1316–C1324. doi: 10.1152/ajpcell.1996.271.4.C1316. [DOI] [PubMed] [Google Scholar]
- PARRATT J.R., VEGH A., PAPP J.G. Bradykinin as an endogenous myocardial protective substance with particular reference to ischemic preconditioning – a brief review of the evidence. Can. J. Physiol. Pharmacol. 1995;73:837–842. doi: 10.1139/y95-114. [DOI] [PubMed] [Google Scholar]
- PITT B., MASON J., CONTI C.R., COLMAN R.W. Activation of plasma kallikrein system during myocardial ischaemia. Adv. Exp. Biol. 1970;8:343–347. [Google Scholar]
- PRUNEAU D., BÉLICHARD P. Induction of bradykinin B2 receptor-mediated relaxation in the isolated rabbit carotid artery. Eur. J. Pharmacol. 1993;239:63–67. doi: 10.1016/0014-2999(93)90976-o. [DOI] [PubMed] [Google Scholar]
- REGOLI D., BARABE J., PARK W.K. Receptors for bradykinin in rabbit aortae. Can. J. Physiol. Pharmacol. 1977;55:855–867. doi: 10.1139/y77-115. [DOI] [PubMed] [Google Scholar]
- REGOLI D., MARCEAU F., LAVIGNE J. Induction of beta 1-receptors for kinins in the rabbit by a bacterial lipopolysaccharide. Eur. J. Pharmacol. 1981;71:105–115. doi: 10.1016/0014-2999(81)90391-5. [DOI] [PubMed] [Google Scholar]
- ROWLAND R.T., CLEVELAND J.C., MENG X., AO L., HARKEN A.H., BROWN J.M. A single endotoxin challenge induces delayed myocardial protection against infarction. J. Surg. Res. 1996;63:193–198. doi: 10.1006/jsre.1996.0246. [DOI] [PubMed] [Google Scholar]
- ROWLAND R.T., MENG X., CLEVELAND J.C., JR, MELDRUM D.R., HARKEN A.H., BROWN J.M. LPS-induced delayed myocardial adaptation enhances acute preconditioning to optimize postischemic cardiac function. Am. J. Physiol. 1997;272:H2708–H2715. doi: 10.1152/ajpheart.1997.272.6.H2708. [DOI] [PubMed] [Google Scholar]
- SONG W., FURMAN B.L., PARRATT J.R. Attenuation by dexamethasone of endotoxin protection against ischaemia-induced ventricular arrhythmias. Br. J. Pharmacol. 1994;113:1083–1084. doi: 10.1111/j.1476-5381.1994.tb17105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORSTILA I. Kallikrein-kinin system in myocardial ischaemia. Acta Med. Scand. 1978;620:1–62. [PubMed] [Google Scholar]
- XI L., JARRETT N.C., HESS M.L., KUKREJA R.C. Essential role of inducible nitric oxide synthase in monophosphoryl lipid A-induced late cardioprotection. Circulation. 1999;99:2157–2163. doi: 10.1161/01.cir.99.16.2157. [DOI] [PubMed] [Google Scholar]
- ZACHAROWSKI K., OTTO M., HAFNER G., CHATTERJEE P.K., THIEMERMANN C. Endotoxin induces a second window of protection in the rat heart as determined by using p-nitro-blue tetrazolium staining, cardiac troponin T release and histology. Arterioscler. Thromb. Vasc. Biol. 1999;19:2276–2280. doi: 10.1161/01.atv.19.9.2276. [DOI] [PubMed] [Google Scholar]
- ZHAO L., WEBER P.A., SMITH J.R., COMERFORD M.L., ELLIOTT G.T. Role of inducible nitric oxide in pharmacological “preconditioning” with monophosphoryl lipid A. J. Mol. Cell. Cardiol. 1997;29:1567–1576. doi: 10.1006/jmcc.1997.0390. [DOI] [PubMed] [Google Scholar]