

Abstract
Spiroxatrine was identified as a moderately potent (Ki=118 nM) but non-selective agonist at the human nociceptin/orphanin FQ receptor, ORL1. This compound was subject to chemical modification and one of the resulting compounds, (8-naphthalen-1-ylmethyl-4-oxo-1-phenyl-1,3,8-triaza-spiro[4.5]dec-3-yl)-acetic acid methyl ester (NNC 63-0532) was shown to have high affinity for ORL1 (Ki=7.3 nM).
NNC 63-0532 showed only moderate affinity for the following receptors (Ki values in parentheses): μ-opioid (140 nM), κ-opioid (405 nM), dopamine D2S (209 nM), dopamine D3 (133 nM) and dopamine D4.4 (107 nM) out of 75 different receptors, ion-channels and transporters.
In functional assays, NNC 63-0532 was shown to be an agonist at ORL1 (EC50=305 nM), a much weaker agonist at the μ-opioid receptor (EC50>10 μM) and an antagonist or weak partial agonist at dopamine D2S (IC50=2830 nM).
Thus, NNC 63-0532 is a novel non-peptide agonist with ∼12 fold selectivity for ORL1 and may be useful for exploring the physiological roles of this receptor owing to its brain-penetrating properties.
Keywords: Nociceptin receptor, ORL1, Orphanin FQ, opioid receptor spiroxatrine
Introduction
Opioid receptors consist of a family of G-protein coupled receptors termed μ, δ, κ and the more recently discovered nociceptin receptor (also termed orphan opioid receptor-like (ORL1)) in case of the human homologue. Nociceptin, also known as orphanin FQ, has been identified as a naturally occurring agonist of ORL1 and is a heptadecapeptide structurally similar to dynorphin A, but lacking the N-terminal tyrosine essential for activation of traditional (μ,κ,δ) opioid receptors (Meunier et al., 1997). Nociceptin has been frequently used to study the physiological roles of ORL1 and it is now clear that this receptor is implicated in a variety of functions in the periphery (e.g., vascular contraction, control of heart rate, water retention, pain perception) and in brain (e.g., memory and learning, pain sensation, control of appetite) (Darland et al., 1998; Meunier et al., 1997). However, in brain and periphery these studies are complicated by the labile nature of nociceptin in vivo and in studies of its central effects by the inability of nociceptin to cross the blood brain barrier. Selective non-peptide ORL1 agonists and antagonists would therefore be highly advantageous in such studies. Very recently, a high affinity and selective ORL1 antagonist, (1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one) has been described (Kawamoto et al., 1999) but only a few non-peptide ORL1 agonists have been reported including lofentanil (Ki=24 nM), etorphine (Ki=530 nM) (Butour et al., 1997) and buprenorphine (IC50=8.4 nM) (Wnendt et al., 1999). Unfortunately, all of these compounds display a quite unfavourable selectivity ratio for ORL over other opioid receptors such as the μ-opioid receptor (their respective Ki's for the μ-opioid receptor are 0.023 nM, 0.18 nM and 0.51 nM (Maguire et al., 1992; Raynor et al., 1995)). In the present study, we discovered that the selective 5-HT1A agonist, spiroxatrine, also showed moderate affinity for ORL1 (Ki=142 nM) and by chemical modification we were able to reverse its selectivity ratio in favour of ORL1 with NNC 63-0532 which is a potent ORL1 agonist (Ki=7.3 nM) with at least 12 fold selectivity over related receptors such as the human μ-opioid receptor.
Methods
The synthesis of NNC 63-0532 contained in a library of 8-substituted 4-oxo-1-phenyl-1,3,8-triaza-spiro[4,5]decanes was performed in a parallel fashion on a solid support as described previously (Watson et al., 1999). In brief, (8-Fmoc-4-oxo-1-phenyl-1,3,8-triazaspiro[4,5]dec-3-yl)ace-tic acid (Fmoc-CPTD-OH, Chem-Impex Int. Inc., Wood Dale, U.S.A.) was attached to Wang-resin using the coupling procedure described by Sieber et al., (1987). The Fmoc protecting group was removed by treatment of the loaded resin with 20% piperidine in N,N-dimethylformamide for 0.5 h. The 8-alkylated derivatives were obtained by a reductive amination procedure in two steps by addition of the appropriate aldehyde, here naphthalene-1-carboxaldehyde, in a mixture of tetrahydrofuran and acetic acid and subsequent reduction of the iminium intermediates with sodium cyanoborohydride. The products were cleaved from the solid support with sodium methoxide/methanol to afford the respective methyl esters. Pure NNC 63-0532 (99% as determined by HPLC and LC/MS) was obtained by recrystallization from ethyl acetate. The Log P value of NNC 63-0532 was determined to 4.32 as the partition coefficient between octanol and phosphate buffer at pH 7.4. Spiroxatrine, [D-Ala2, N-Me-Phe4, Gly-ol5]-enkephalin (DAMGO) and morphine were obtained from Sigma-Aldrich, St. Louis, U.S.A. Nociceptin and the hexapeptide, ac-RYYRWK-NH2 (Dooley et al., 1997) were custom synthesized at Novo Nordisk A/S, Denmark.
Membrane preparation
BHK cells expressing ORL1 in a pcDNA 3.1 (In Vitrogen, Carlsbad, CA, U.S.A.) were harvested with phosphate buffered saline supplemented with 1 mM EDTA, pH 7.4 and centrifuged (1000×g, 5 min, 4°C). The pellet was homogenized in assay buffer (50 mM TRIS-HCl, pH 7.8, 5 mM MgCl2, 0.2 mM EGTA, 1 mg ml−1 BSA, 0.2 mg ml−1 bacitracin), centrifuged, suspended in buffer and frozen in aliquots at −80°C.
[125I]-Tyr14-Nociceptin binding assay to human ORL1
[125I]-Tyr14-Nociceptin binding was performed using a modification of a previously described protocol (Ardati et al., 1997). In brief, frozen membranes were thawed, homogenized briefly and kept on ice until use. For competition binding studies, test compounds, membranes (0.05 mg protein per well), 1.0 mg SPA beads per well (WGA PVT beads, Amersham, Buckinghamshire, U.K.) and assay buffer (see above) was added to 96 well plates at 25°C in a final volume of 200 μl. The incubation (120 min at 25°C) was started by adding 0.05 nM [125I]-Tyr14-Nociceptin (New England Nuclear, Boston, U.S.A., specific activity=2200 Ci mmol−1) and plates were counted in a Top-counter (Packard, Illinois, U.S.A) after settling of the beads. Non-specific binding was defined as binding in the presence of 1 μM nociceptin and accounted for 4–8% of total binding.
[35S]-Guanosine-5′-O-(3-thio)triphosphate-γ-S ([35S]-GTP-γ-S) binding assays
The functional activity of ORL1 ligands were determined using a [35S]-GTP-γ-S binding assay essentially as described previously (Albrecht et al., 1998). In brief, test compounds, membranes (0.1 mg protein per well), 2.0 mg SPA beads, 1 μM guanosine-diphosphate (Sigma, St. Louis, U.S.A.) and assay buffer (composition: 50 mM TRIS-HCl, pH 7.8, 100 mM NaCl, 5 mM MgCl2, 0.2 mM EGTA, 1 mg/ml BSA, 0.2 mg/ml bacitracin) was added to 96 well plates at 25°C in a volume of 175 μl. After 30 min pre-incubation at 25°C, the incubation (60 min at 25°C) was started by adding [35S]-GTP-γ-S (New England Nuclear, Boston, U.S.A., specific activity=1100 Ci mmol−1) to a final concentration of 0.2 nM. Plates were counted in a Top-counter after settling of the beads. Maximal stimulation to nociceptin was determined from dose-response curves and was 50–80% above basal levels of [35S]-GTP-γ-S binding.
Measurements of cyclic AMP (cAMP)-formation
Measurements of cyclic AMP-formation in BHK cells expressing ORL1 were performed essentially as previously described (Thomsen et al., 1992). Briefly, the cells were seeded in 24-well plates two days before the experiment and then washed once with cyclic AMP-assay buffer (composition (mM): NaCl 118, MgSO4 1.2, CaCl2 2.5, KH2PO4 1.2, KCl 4.7, glucose 11, NaHCO3 25, HEPES 10, pH 7.4 and 3-isobutyl-1-methylxanthine 1). The cells were incubated with test compounds just prior to stimulation of adenylate cyclase with 10 μM forskolin (Calbiochem, U.S.A.) for 10 min at 37°C. The incubation was stopped by adding 25 μl HCl (2 M) and the cyclic AMP contents in 25 μl aliquots were determined using a radioimmuno assay kit (Amersham, Buckinghamshire, U.K.).
Determination of extracellular acidification rates from dopamine D2S cells
The Cytostar microphysiometer (Molecular Devices, Menlo Park, CA, U.S.A.) was used for obtaining functional measurements of D2S activation because the receptor was not coupled efficiently to inhibition of cyclic AMP-formation in the cell line studied (data not shown). These measurements were performed as described previously (Scheideler et al., 1997). Briefly, dopamine D2S expressing cells were seeded into cytosensor microphysiometer cups (3×105 cells per cup) 18–24 h before the experiments were initiated. Flow chambers containing the cells were perfused at a rate of 100 μl min−1 with a running buffer composed of bicarbonate-free DMEM (pH 7.4) containing 44 mM NaCl. The total pump cycle had a duration of 2 min with 80 s at a flow rate of 100 μl min−1 and 40 s with the flow stopped. The changes in extracellular pH was measured for 30 s during each cycle while the flow was stopped and automatically converted to acidification rates (CytoSoft program, Molecular Devices, Menlo Park, CA, U.S.A.). Test compounds were diluted into the running buffer and pH was adjusted to 7.4. A recovery period of at least 20 min was allowed between drug exposures to allow for the stabilization of acidification rates. When testing for antagonist effects, the compounds were applied 5 min before the agonist e.g., 30 nM quinpirole (Sigma-Aldrich, St. Louis, U.S.A.).
Miscellaneous receptor binding assays
NNC 63-0532 was submitted to a high throughput receptor profile (CEREP, Celle l'Evescault, France) and tested according to the protocols described by the company (1998 catalogue) for activity at 1 μM at the receptors mentioned in the Results section. [3H]-DAMGO bindings were performed as described (Raynor et al., 1995) using 0.06 mg of human μ-opioid receptor expressing membranes (Receptor Biology Inc., Beltsville, U.S.A.) and 1 nM [3H]-DAMGO (New England Nuclear, Boston, U.S.A., specific activity=55 Ci mmol−1) for displacement assays. [3H]-Spiperone binding to human D2S, D3 and D4.4 was performed essentially as described in Scheideler et al. (1997) using 0.05 mg per assay point of the respective receptor expressing membranes and 0.1 nM (D2S) or 0.3 nM (D3 and D4.4) [3H]-spiperone (New England Nuclear, Boston, U.S.A., specific activity=15 Ci mmol−1) for displacement assays. [3H]-(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxa-spiro[4.5]dec-8-yl]-benzeneacetamide ([3H]U-69593) binding was performed as described (Raynor et al., 1994) using 0.1 mg of human κ-opioid receptor expressing membranes (Receptor Biology Inc., Beltsville, U.S.A.) and 1 nM [3H]-U-69593 (New England Nuclear, Boston, U.S.A., specific activity=49 Ci mmol−1) for displacement assays. [3H]-Diprenorphine binding to rat cortical membranes was performed essentially as detailed previously (Rodriguez et al., 1992) using 1.0 mg rat cortical membranes (P2-fraction) in assay buffer (25 mM HEPES, pH 7.4+10 mM MgCl2) which were incubated for 60 min at 25°C with 0.5 nM [3H]-diprenorphine (New England Nuclear, Boston, U.S.A., specific activity=55 Ci mmol−1).
Results
Because lofentanil has been reported to have moderate affinity for ORL1 (Butour et al., 1997) in addition to its pM affinity for μ-opioid receptors (Maguire et al., 1992), a number of structurally related compounds including the 5-HT1A agonist, spiroxatrine, were tested for ORL1 binding affinity. Spiroxatrine showed moderate affinity for ORL1 (Ki=118±16 nM) and was therefore subject to chemical modification aimed at improving its potency and selectivity for ORL1. This led, among other compounds (Watson et al., 1999) to NNC 63-0532 (Figure 1) which showed high affinity for both the human recombinant ORL1 (Ki=7.3±0.9 nM) (Figure 2A) and for the endogenous nociceptin receptor in rat cortical membranes (Ki=11±2 nM) (Figure 2B).
Figure 1.
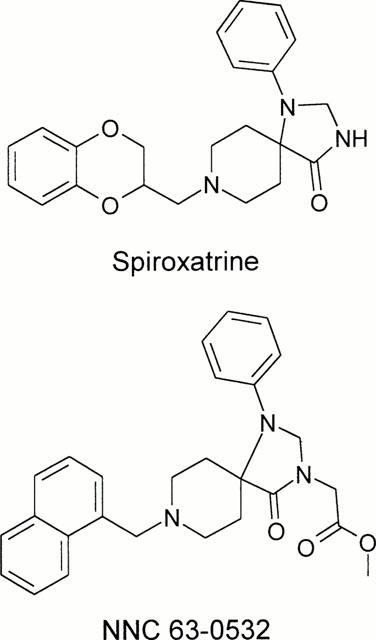
Chemical structure of spiroxatrine and NNC 63-0532.
Figure 2.
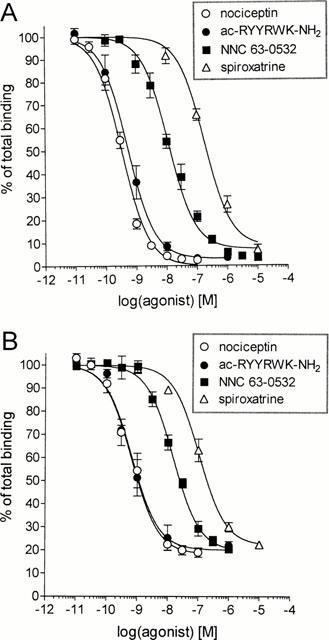
Affinity of NNC 63-0532 and reference compounds at (A) the cloned human ORL1 and B) the rat nociceptin receptor in cortical membranes. The data are mean±s.e.mean of 3–4 experiments, which were performed in triplicate. IC50 values were calculated from competition binding experiments by a non-linear regression analysis fitted to a one-site model using the GraphPad Prism program (GraphPad Software, San Diego, U.S.A.). In separate experiments, the dissociation constants (Kd) for [125]-Tyr14-nociceptin binding to ORL1-expressing cells (Kd=0.10±0.02 nM, n= 4) and rat cortical membranes (Kd=0.15±0.04 nM, n= 3) were determined and used for calculation of Ki values according to the equation: Ki=IC50/(1+[L]/Kd) where [L] is the concentration of radioligand.
In order to verify the selectivity of NNC 63-0532 for ORL1, the compound was profiled in a commercial screen for activity at 75 different receptors, ion-channels and transporters. At a concentration of 1 μM, NNC 63-0532 did not display appreciable affinity for the following targets (less than 50% inhibition at 1 μM): adenosine (A1–A3), α1- and α2-adrenergic, β1- and β2-adrenergic, norepinephrine uptake, angiotensin 1 and 2, atrial natriuretic peptide, central and peripheral benzodiazepine receptors, bombesin, bradykinin B2, calcitonin gene related peptide, cannabinoid CB1 and CB2, cholecystokinin A and B, dopamine D1 and D5, dopamine uptake, endothelin A and B, GABA (non-selective), galanin, platelet derived growth factor, interleukin 1b, tumour necrosis factor α, chemokine receptor 1, histamine H1, melatonin receptor 1, muscarinic (m1–m5), tachykinin NK2 and NK3, neuropeptide Y1 and Y2, neurotensin, pituitary adenylate cyclase-activating polypeptide, non-competitive N-methyl-D-aspartate site, prostanoid thromboxane A2, prostaglandin I2, purine P2X and P2Y, serotonin receptors (5-HT1A, 5-HT1B, 5-HT2A, 5-HT2C, 5-HT3, 5-HT5A, 5-HT6, 5-HT7), somatostatin, vasoactive intestinal peptide, vasopressin V1A, Ca2+-channel (L-type), voltage dependent potassium channels or Na+- and Cl−-channels. NNC 63-0532 showed weak to moderate activity (50–70% inhibition) for the non-selective σ site, histamine H2 receptor, δ-opioid receptor and the tachykinin NK1 receptor. NNC 63-0532 did show moderate to high activity (70–90% inhibition) for μ- and κ-opioid receptors and for dopamine D2, D3 and D4 receptors. Accordingly, the activity of NNC 63-0532 for dopamine and opioid receptors was explored in more detail. As shown in Figure 3A, NNC 63-0532 showed about 20 fold selectivity for ORL1 when measuring displacement of radioligand binding to human ORL1, human μ-opioid and human κ-opioid receptors (respective Ki values were 7.3±0.9 nM, 140±22 nM and 405±54 nM). The selectivity of NNC 63-0532 for ORL1 was about 14 fold when measuring displacement of radioligand binding to dopamine D2S, D3 and D4.4 receptors (respective Ki values were 209±32 nM, 133±14 nM and 107±9 nM) (Figure 3B). To examine the activity of NNC 63-0532 for rat opioid receptors the non-selective opioid radioligand, [3H]-diprenorphine, was used in displacement binding studies with rat cortical membranes. NNC 63-0532 displaced [3H]-diprenorphine binding only at 50 fold higher concentrations (Ki=560±55 nM, Figure 4) than its affinity for the rat nociceptin receptor (Figure 2B). In these experiments, morphine showed a bi-phasic displacement curve with a high affinity site (Ki=0.7±0.1 nM) and a lower affinity site (Ki=64±15 nM) in agreement with its non-selective action at opioid receptors (Raynor et al., 1994). In functional assays, NNC 63-0532 was shown to be an agonist with an efficacy which was close to that of nociceptin both when measuring inhibition of cyclic AMP-formation in BHK cells expressing ORL1 (98±3%, Figure 5A) or when measuring stimulation of [35S]-GTP-γ-S binding to membranes from these cells (95±4%, Figure 5B).
Figure 3.
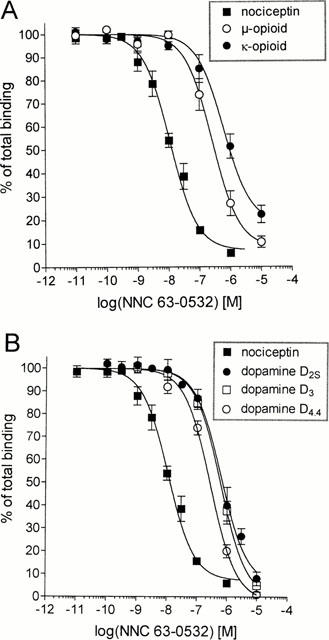
(A) Selectivity of NNC 63-0532 at human ORL1, μ-opioid and κ-opioid receptors, and (B) Selectivity of NNC 63-0532 for human ORL1 over dopamine D2S, D3 and D4.4 receptors. The resulting IC50's were converted to Ki values (see Results) as described in the legend to Figure 2 using the following parameters: ORL1 (Kd=0.10 nM; [L]=0.05 nM); μ-opioid (Kd=1.2 nM; [L]=1 nM); κ-opioid (Kd=2.1 nM; [L]=1 nM); dopamine D2S (Kd=0.05 nM; [L]=0.1 nM); dopamine D3 (Kd=0.09 nM; [L]=0.3 nM) and dopamine D4.4 (Kd=0.22 nM; [L]=0.3 nM).
Figure 4.
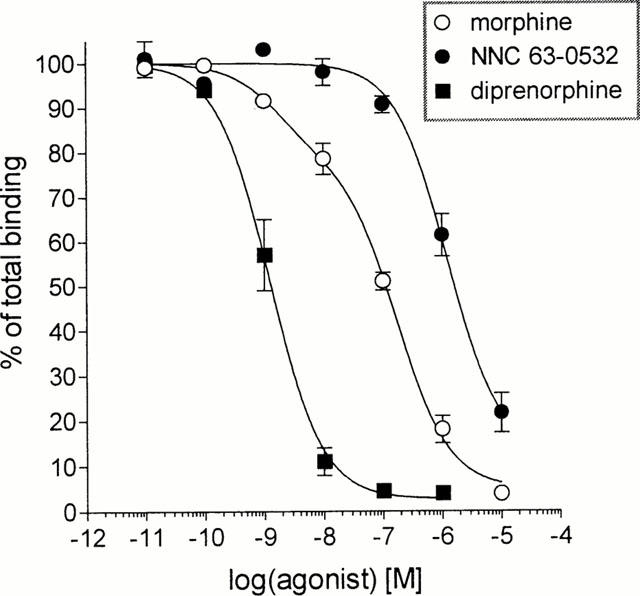
Selectivity of NNC 63-0532 for the rat nociceptin receptor over rat opioid receptors as measured by displacement of [3H]-diprenorphine binding by NNC 63-0532, morphine and diprenorphine. The data are the mean±s.e.mean of three experiments, which were performed in triplicate. The displacement curves were compared to, respectively, one- and two-site competition binding models using least square analysis with GraphPad Prism software. Only the displacement curve for morphine was preferentially fitted to a two-site competition model over a one-site model (P<0.001). For Ki values see text.
Figure 5.
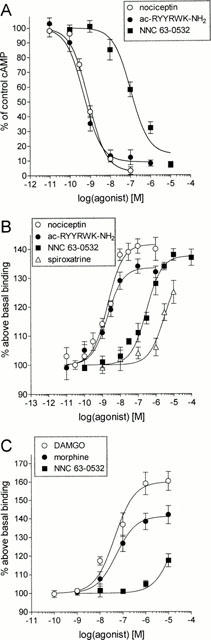
Functional agonism of NNC 63-0532 at ORL1 when measuring (A) inhibition of forskolin-induced cyclic AMP-formation in BHK cells expressing ORL1, (B) stimulation of [35S]-GTP-γ-S binding to membranes from BHK/ORL1 cells and (C) stimulation of [35S]-GTP-γ-S binding to membranes from human μ-opioid receptor expressing cells. The calculated EC50's were (A) 0.83±0.15 nM (nociceptin), 0.49±0.08 nM (ac-RYYRWK-NH2) and 109±11 nM (NNC 63-0532); (B) 2.0±0.1 nM (nociceptin), 1.9±0.2 nM (ac-RYYRWK-NH2), 305±26 nM (NNC 63-0532) and 3050±210 nM (spiroxatrine) and (C) 44±6 nM (DAMGO), 52±8 nM (morphine) and >10.000 nM (NNC 63-0532). The results are mean±s.e.mean of 3–4 separate experiments that were performed in triplicate.
The functional activity of NNC 63-0532 was also determined at human μ-opioid receptors measuring stimulation of [35S]-GTP-γ-S binding. As shown in Figure 5C, NNC 63-0532 was a weak agonist at μ-opioid receptors with an EC50 in excess of 10 μM. Because the dose-response curve did not reach saturation at the highest concentration which could be applied within the limit of solubility it was not possible to determine the actual potency or relative efficacy of NNC 63-0532 at the μ-opioid receptor. At the dopamine D2S receptor, NNC 63-0532 did not elicit functional responses per se in the microphysiometer assay (Scheideler et al., 1997) but antagonized sub-maximal stimulation of the acidification rate evoked by 30 nM quinpirole with low potency (IC50=2830±490 nM, n= 3) (data not shown). Thus, NNC 63-0532 appears to be an antagonist or a weak partial agonist at the dopamine D2S receptor.
Discussion
Despite the fact that much information regarding the central physiological roles of ORL1 has been obtained using local infusion of the endogenous heptadecapeptide, nociceptin, the availability of potent and selective non-peptide ORL1 agonists is likely to greatly facilitate the understanding of the functioning of this receptor and its therapeutic relevance. In the present study, a new potent agonist at ORL1, NNC 63-0532 has been characterized and shown to be at least 12 fold selective for ORL1 in terms of binding affinity for the human receptors. While its parent compound, spiroxatrine, is a nM potent 5-HT1A agonist, NNC 63-0532 showed very low affinity for 5-HT1A. By preparing a number of compounds structurally related to NNC 63-0532 we observed that a naphthyl group in the 8-position was optimal for obtaining high affinity and selectivity for ORL1 (Watson et al., 1999). The affinity of NNC 63-0532 for ORL1 is about 10 fold lower as compared to nociceptin but almost similar to buprenorphine's ORL1 affinity (Wnendt et al., 1999). However, when compared to previously described non-peptide agonists acting at ORL1 such as lofentanil and buprenorphine which have selectivity ratios (expressed as μ-opioid/ORL1 Ki-ratios) of 0.001 and 0.06, respectively the similar ratio for NNC 63-0532 (∼20) is much more favourable. In agreement with previous observations (Butour et al., 1997; Dooley et al., 1997), nociceptin and ac-RYYRWK-NH2 inhibited forskolin-induced cyclic AMP-formation in ORL1 expressing cells with a similar high nM potency. However, ac-RYYRWK-NH2 was a full agonist in the cyclic AMP assay, which is in contradiction to the previously observed partial agonism found in a similar assay for the mouse ORL1 (Dooley et al., 1997). This discrepancy may be related to the presence of spare receptors (Kenakin, 1997) in the cell line used in the present study. We therefore employed a [35S]-GTP-γ-S binding assay which is well suited for detecting eventual partial agonism (see Lazareno, 1999 for review). In such experiments, the efficacy of NNC 63-0532 persisted to be very high (i.e., 95% relative to nociceptin) whereas ac-RYYRWK-NH2 appeared as a partial agonist in agreement with previous observations (Dooley et al., 1997). Thus, NNC 63-0532 is an agonist at ORL1 with an efficacy almost comparable to that of nociceptin at least in the functional assays used in the present study. The potencies of all agonists tested in the functional ORL1 assays were lower when compared to their respective binding affinities. Similar observations have been made previously with ORL1 (Adapa & Toll, 1997; Albrecht et al., 1998; Butour et al., 1997; Dooley et al., 1997)) and with several other G-protein coupled receptors (Kenakin, 1997). This is most likely related to the different assay conditions (e.g., presence/absence of GTP and Na+-ions) which largely affects the apparent potency of nociceptin agonists (Adapa & Toll, 1997; Albrecht et al., 1998; Ardati et al., 1997). Under physiological conditions sufficient GTP will be present to allow for only the low affinity site to be detected (for review see: Waelbroeck, 1999). However, NNC 63-0532 was also much less potent in the functional μ-opioid assay as compared to its affinity for displacing [3H]-DAMGO binding and therefore, the relative selectivity of NNC 63-0532 for ORL1 over μ-opioid receptor was also evident when compared in functional assays. Perhaps not unexpected from the high log P value of NNC 63-0532 (Log P was 4.32), initial behavioural studies have shown that NNC 63-0532 induces effects that are consistent with central ORL1 activation upon peripheral (s.c.) administration (facilitation of lordosis behaviour and depression of locomotor activity) (De Beun, unpublished information), indicating that the compound does cross the blood brain barrier.
In conclusion, the present study shows that a nM potent non-peptide agonists with full efficacy at ORL1 can be prepared based on chemical modification of the 5HT1A agonist, spiroxatrine. This compound, NNC 63-0532 represents a significant progress towards the development of entirely selective non-peptide ORL1 agonists and may prove useful for further exploring the physiology of ORL1 and its therapeutic possibilities.
Abbreviations
- BHK
baby hamster kidney
- DAMGO
[D-Ala2, N-Me-Phe4, Gly-ol5]-enkephalin
- GTP-γ-S
guanosine-5′-O-(3-thio)trisphosphate-γ-S
- NNC 63-053
(8-naphthalen-1-ylmethyl-4-oxo-1-phenyl-1,3,8-triaza-spiro[4.5]dec-3-yl)-acetic acid methyl ester
- ORL1
orphan opioid receptor-like
- U 69593
(+)−(5α,7α,8β)-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
A non-peptide nociceptin agonist with structural similarities to NNC 63-0532 and anxiolytic activity in rodents has been reported (Jenck et al., 2000, Proc. Natl. Acad. Sci. U.S.A., 97, 4938–4943).
References
- ADAPA I.D., TOLL L. Relationship between binding affinity and functional activity of nociceptin/orphanin FQ. Neuropeptides. 1997;31:403–408. doi: 10.1016/s0143-4179(97)90032-9. [DOI] [PubMed] [Google Scholar]
- ALBRECHT E., SAMOVILOVA N.N., OSWALD S., BAEGER I., BERGER H. Nociceptin (orphanin FQ): high-affinity and high capacity binding site coupled to low potency stimulation of guanylyl-5′-o-(γ-thio)-triphosphate binding in rat brain membranes. J. Pharmacol. Exp. Ther. 1998;286:896–902. [PubMed] [Google Scholar]
- ARDATI A., HENNINGSEN R.A., HIGELIN J., REINSCHEID R.K., CIVELLI O., MONSMA F.J. Interaction of [3H]orphanin FQ and 125I-Tyr14-orphanin FQ with the orphanin FQ receptor: kinetics and modulation by cations and guanine nucleotides. Mol. Pharmacol. 1997;51:816–824. doi: 10.1124/mol.51.5.816. [DOI] [PubMed] [Google Scholar]
- BUTOUR J.L., MOISAND C., MAZARGUIL H., MOLLEREAU C., MEUNIER J.C. Recognition and activation of the opioid receptor-like ORL 1 receptor by nociceptin, nociceptin analogs and opioids. Eur. J. Pharmacol. 1997;321:97–103. doi: 10.1016/s0014-2999(96)00919-3. [DOI] [PubMed] [Google Scholar]
- DARLAND T., HEINRICHER M.M., GRANDY D.K. Orphanin FQ/nociceptin – a role in pain and analgesia, but so much more. Trends Neurosci. 1998;21:215–221. doi: 10.1016/s0166-2236(97)01204-6. [DOI] [PubMed] [Google Scholar]
- DOOLEY C.T., SPAETH C.G., BERZETEIGURSKE I.P., CRAYMER K., ADAPA I.D., BRANDT S.R., HOUGHTEN R.A., TOLL L. Binding and in vitro activities of peptides with high affinity for the nociceptin/orphanin FQ receptor, ORL1. J. Pharmacol. Exp. Ther. 1997;283:735–741. [PubMed] [Google Scholar]
- KAWAMOTO H., OZAKI S., ITOH Y., MIYAJI M., ARAI S., NAKASHIMA H., KATO T., OHTA H., IWASAWA Y. Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: (1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one) (J-113397) J. Med. Chem. 1999;25:5061–5063. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Differences between natural and recombinant G protein-coupled receptor systems with varying receptor/G protein stoichiometry. Trends. Pharmacol. Sci. 1997;18:456–464. doi: 10.1016/s0165-6147(97)01136-x. [DOI] [PubMed] [Google Scholar]
- LAZARENO S. Measurement of agonist-stimulated [35S]GTP-γ-S binding to cell membranes. Methods Mol. Biol. 1999;106:231–245. doi: 10.1385/0-89603-530-1:231. [DOI] [PubMed] [Google Scholar]
- MAGUIRE P., TSAI N., KAMAI J., COMETTA-MORINI C., UPTON C., LOEW G. Pharmacological profiles of fentanyl analogs at μ, κ and δ opiate receptors. Eur. J. Pharmacol. 1992;213:219–225. doi: 10.1016/0014-2999(92)90685-w. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur. J. Pharmacol. 1997;340:1–15. doi: 10.1016/s0014-2999(97)01411-8. [DOI] [PubMed] [Google Scholar]
- RAYNOR K., KONG H., CHEN Y., YASUDA K., YU L., BELL G.I., REISINE T. Pharmacological characterization of the cloned κ-, δ-, and μ-opioid receptors. Mol. Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- RAYNOR K., KONG H., MESTEK A., BYE L.S., TIAN M., LIU J., YU L., REISINE T. Characterization of the cloned human μ-opioid receptor. J. Pharmacol. Exp. Ther. 1995;272:423–428. [PubMed] [Google Scholar]
- RODRIGUEZ F.D., BARDAJI E., TRAYNOR J.R. Differential effects of Mg2+ and other divalent cations on the binding of tritiated opioid ligands. J. Neurochem. 1992;59:467–472. doi: 10.1111/j.1471-4159.1992.tb09393.x. [DOI] [PubMed] [Google Scholar]
- SCHEIDELER M.A., MARTIN J., HOHLWEG R., RASMUSSEN J.S., NAERUM L., LUDVIGSEN T.S., LARSEN P.J., KORSGAARD N., CRIDER A.M., GHOSH D., CRUSE S.F., FINK-JENSEN A. The preferential dopamine D3 receptor agonist cis-8-OH-PBZI induces limbic Fos expression in rat brain. Eur. J. Pharmacol. 1997;339:261–270. doi: 10.1016/s0014-2999(97)01372-1. [DOI] [PubMed] [Google Scholar]
- SIEBER P. An improved method for anchoring of 9-fluorenylmethoxycarbonyl-amino acids to 4-alkoxybenzyl alcohol resin. Tetrahedron Lett. 1987;28:6147–6150. [Google Scholar]
- THOMSEN C., KRISTENSEN P., MULVIHILL E., HALDEMAN B., SUZDAK P.D. L-2-Amino-4-phosphonobutyrate (L-AP4) is an agonist at the type IV metabotropic glutamate receptor which is negatively coupled to adenylate cyclase. Eur. J. Pharmacol. Mol. Pharmacol. 1992;227:361–363. doi: 10.1016/0922-4106(92)90018-q. [DOI] [PubMed] [Google Scholar]
- WAELBROECK M. Kinetics versus equlibrium: the importance of GTP in GPCR activation. Trends Pharmacol. Sci. 1999;20:477–481. doi: 10.1016/s0165-6147(99)01394-2. [DOI] [PubMed] [Google Scholar]
- WATSON B.T., HOHLWEG R., THOMSEN C.Novel 1,3,8-triazaspiro[4,5]decanones with affinity for opioid receptor subtypes PCT Int. Appl. 1999. WO 9959997
- WNENDT S., KRUGER T., JANOCHA E., HILDEBRANDT D., ENGLBERGER W. Agonistic effect of buprenorphine in a nociceptin/OFQ receptor-triggered reporter gene assay. Mol. Pharmacol. 1999;56:334–338. doi: 10.1124/mol.56.2.334. [DOI] [PubMed] [Google Scholar]