

Abstract
The vasoactive peptide endothelin (ET) has been implicated in the pathogenesis of cerebral vasospasm following subarachnoid haemorrhage. In these studies we investigated the involvement of protein kinase C (PKC) in sustained vasoconstriction induced by ET-1 in canine cerebral arteries. We also examined the ability of the aminoglycoside antibiotics to reverse the effects mediated by ET-1 in canine cerebrovascular smooth muscle cells (CVSMC).
The ETA receptor antagonist, BQ-123, showed a competitive inhibition of the ET-1 responses.
The vasoconstrictor action of both ET-1 (0.5 nM) and phorbol myristate acetate (PMA) (160 nM) was reversed by a selective PKC inhibitor, Ro-32-0432.
In cerebral arteries precontracted with ET-1 the aminoglycosides caused a concentration-dependent relaxation. The EC50s for the relaxation were as follows: 0.54±0.05, 0.63±0.01, 1.88±0.46 and 2.3±0.92 mM for gentamicin, neomycin, streptomycin and kanamycin, respectively.
Gentamicin caused a concentration-dependent decrease of the PMA-induced responses in calcium free medium.
PKC activity was elevated in CVSMC exposed to ET-1 (170%) and PMA (167%) for a period of time (60 min) corresponding to maximum tonic contraction induced by these agents in arterial rings.
The administration of the aminoglycosides to CVSMC, in concentrations corresponding to the EC50s from contractility studies, reduced the effects of both ET-1 and PMA on PKC activity to the levels not different from controls.
These results show that the aminoglycosides are able to inhibit sustained vasoconstriction induced by ET-1, an effect which is due, at least in part, to the inhibition of PKC.
Keywords: Cerebral vasospasm, endothelin, aminoglycosides, protein kinase C
Introduction
Delayed cerebral vasospasm remains one of the major causes of morbidity and mortality in patients with subarachnoid haemorrhage (SAH), and at present there is no satisfactory treatment which can prevent or reverse cerebral vasoconstriction in humans (Cook & Vollrath, 1995). Although the pathogenesis of this condition is unclear, there is evidence that one of the principal causative agents in vasospasm is endothelin-1 (ET-1), a potent vasoactive peptide generated in vascular endothelial and smooth muscle cells (Zimmermann & Seifert, 1998). Levels of ET-1 are elevated in the cerebrospinal fluid (CSF) of patients with SAH, and the size of these changes is correlated with the timing and severity of the spasm (Seifert et al., 1995). In addition, the ETA receptor antagonists, BQ-123 (Itoh et al., 1994; Josko et al., 1998) and TBC 11251 (Wanebo et al., 1998), attenuate cerebral vasospasm in animal models, although not all studies have been positive (Hino et al., 1995). Further evidence in favour of the concept of a role for ET-1 is provided by the observations that synthesis of this peptide is stimulated at the level of gene transcription by a number of factors, including the vasoactive agents, haemoglobin, thrombin and prostaglandin F2α, released from the blood clot after SAH (Schini et al., 1989; Ohlstein & Storer, 1992; Kasuya et al., 1993). Together these findings support the concept that release of ET-1 may play a role in the development of vasospasm, and suggest that inhibition of the effects of the vasoconstrictors released from the blood clot following SAH could provide a valuable means of therapeutic intervention.
The molecular mechanism of ET-mediated sustained cerebral vasoconstriction remains unclear. In vascular smooth muscle this peptide interacts with ETA receptors which are coupled via Gq protein to multiple cellular effectors, including phospholipases C (PLC) and D (PLD), protein kinase C (PKC), tyrosine kinases (Schiffrin & Touyz, 1998; Goldie, 1999), and several classes of calcium channels (Nakajima et al., 1996; Iwamuro et al., 1999).
It has been suggested that a major role in the intracellular events leading to sustained contraction is played by PKC and its serine/threonine phosphorylated substrates (Horowitz et al., 1996). The concept of a role for PKC in cerebral vasospasm is supported by the observations that phorbol esters, potent activators of PKC, induce angiographic vasospasm (Sako et al., 1993), and that the PKC inhibitors improve the outcome in experimental models of cerebral vasospasm (Minami et al., 1992). Furthermore, the level of DAG exhibits a prolonged increase during experimental cerebral vasospasm, suggesting a critical involvement of PKC (Asano & Matsui, 1999). Since ET-1 activates both PKC and PLD in a variety of cells (Eskildsen-Helmond et al., 1997), it is conceivable that sustained cerebral vasoconstriction induced by this peptide is due, at least in part, to a prolonged generation of DAG and thus activation of PKC. We have recently shown that cerebral vasoconstriction induced by haemoglobin, released from haemolyzed blood after SAH, and prostaglandin F2α, can be inhibited by neomycin, a nonselective inhibitor of PLC which has been widely used to assess the involvement of inositol 1,4,5 trisphosphate (IP3) in cellular responses (Gergawy et al., 1998). A number of studies have shown that in addition to PLC, neomycin inhibits other targets, including PKC (Hagiwara et al., 1988), PLD (Rumenapp et al., 1997) and a variety of calcium channels (Perrier et al., 1992; Langton et al., 1996; Pichler et al., 1996). The ability of neomycin and related agents to inhibit a wide range of intracellular events, some of which probably contribute to the vasospastic activity by means of their involvement in the signalling pathways activated by vasoconstrictors, suggests that these compounds, or agents with similar characteristics, could be of potential benefit in the treatment of cerebral vasospasm. Therefore, the present studies were conducted to explore whether PKC may be involved in a sustained vasoconstriction induced by ET-1 in cerebral arteries, and to determine whether the structurally related aminoglycoside antibiotics, gentamicin, neomycin, kanamycin and streptomycin, affect the contractile action of ET-1 and, if so, what mechanism might be involved in this action.
Methods
Recording of isometric tension
Mongrel dogs of either sex, weighing about 20 kg, were used in these studies. Protocols for the humane treatment of animals, according to the Declaration of Helsinki, and as approved by the University of Alberta Animal and Ethics review Committee were followed in all experiments. The animals were killed with an intravenous (i.v.) overdose of pentobarbitone, and the brain with cerebral arteries attached was removed and placed in oxygenated Krebs-Henseleit solution of the following composition (in mM): Na+ 130, K+ 5, Ca2+, 2.5, Mg2+, 1.2, Cl− 12.2, HCO3 25, SO42− 1.2, H2PO42− 1.2 and dextrose 11. Each artery was cut into sections about 3 mm in length and these preparations were suspended in tissue baths containing Krebs-Henseleit solution at 37°C, gassed with 95% oxygen and 5% carbon dioxide. The arterial rings were equilibrated for 1 h. Isometric tension was measured using a strain gauge transducer, attached to a preamplifier and recorded on a potentiometric chart recorder. The resting tension was set at 1.0 g, which is optimal for inducing maximum contraction, and the preparations were allowed to equilibrate for 60 min, during which time the bathing medium was renewed every 15 min. After the equilibration period, contractile responses of the ring preparations to 60 mM KCl were induced. The responses were recorded isometrically using force displacement transducers and a Grass 7D polygraph (Vollrath et al., 1994). To obtain concentration responses to ET cumulative concentrations of these agents were added to organ baths as soon as the maximum response to the preceding concentration had been obtained. In the experiments in which the effects of the aminoglycoside antibiotics, gentamicin, neomycin, kanamycin and streptomycin were measured, the increasing cumulative concentrations of these agents were added after a plateau tension in response to 0.5 nM ET-1 or 160 nM of phorbol-12-myristate-13-acetate (PMA) had developed. The relaxation of the preparations exposed to the aminoglycosides was expressed as the percentage of maximum tension produced by the vasoconstrictors. Similar protocol was employed in the experiments in which the effects of PKC inhibitor, Ro-32-0432, were examined. The presence of an intact endothelium was examined by evaluating the relaxant responses to bradykinin, in an arterial preparation that was precontracted with 1 μM 5-hydroxytryptamine (5-HT). In addition, the relaxing responses to the aminoglycoside antibiotics were analysed in the presence of nitric oxide synthase (NOS) inhibitor (L-NAME). In some experiments, the aminoglycosides (5 mM) were added 30 min before the exposure of cerebral artery preparations to the agonists. The relaxation of the preparations exposed to the aminoglycosides was expressed as the percentage of maximum tension produced by the vasoconstrictors. The EC50 values for the aminoglycosides were determined by fitting an exponential curve to the data using Microsoft Excel.
Cerebrovascular smooth muscle culture
Cells were prepared as described previously (Vollrath et al., 1995). Briefly, canine basilar arteries were isolated under sterile conditions and placed in a Petri dish containing Dulbecco's modified Eagle's medium (DMEM). The adventitia was removed mechanically, the vessels were cut into segments approximately 5 mm in length along the longitudinal axis, and the endothelium was removed by gently scraping the inner surface of the segments. The tissue was then chopped into 1 to 2 mm pieces and the explants were transferred to 25 cm2 culture flasks containing 1 ml of DMEM supplemented with 10% foetal calf serum, penicillin (100 u ml−1) and streptomycin (100 μg ml−1). After the explants had adhered, the volume was made up gradually over the next 4 days to 4 ml per flask, and thereafter the culture medium was changed weekly. When the primary cultures were almost confluent the cerebrovascular smooth muscle cells (CVSMC) were transferred to 75 cm2 flasks and then routinely subcultured at a split ratio of 1 : 3.
Measurements of PKC activity
PKC activity was measured in the membrane and cytosolic fractions of serum-starved and antibiotics (penicillin and streptomycin) free (48 h) CVSMC exposed to ET-1 (0.5 nM) or PMA (160 nM) for 60 min. The aminoglycoside antibiotics were added 30 min after initial exposure to the agonists, in the concentrations equal to the EC50s calculated from contractility studies. After incubation, the cells were scraped in ice-cold phosphate buffered saline (PBS) (145 mM NaCl and 10 mM sodium phosphate; pH 7.4), and centrifuged at 200×g for 10 min. The pelleted cells were homogenized in ice-cold 25 mM Tris-HCl buffer (pH 7.2) containing (mM) EGTA 4, EDTA 2, dithiothreitol 2.5 and leupeptin 20 μM. Homogenized cells were then separated into cytosolic and particulate fractions by centrifugation at 15,000×g for 60 min at 4°C. The supernatant was then assayed for soluble PKC activity which was measured using a PKC enzyme assay system (Pierce). The membrane pellets were resuspended in the homogenization buffer and the PKC was solubilized via sonication in ice-cold homogenization buffer containing 0.4% Triton X-100. The standard reaction mixture (15 μl) contained Tris at pH 7.4 100 (mM), ATP 10 mM, MgCl2 50 mM, CaCl2 0.5 mM, 0.01% Triton X-100, phosphatidylserine (PS) (1 mg ml−1), PMA (1.6 μM), the peptide substrate (pseudosubstrate peptide labelled with a fluorescent dye), and a sample containing the endogenous PKC (10 μl). The samples were incubated at 30°C for 30 min. The reaction mixture was then applied to the separation units containing the affinity membranes (Pierce), which specifically bind the phosphorylated peptide. The bound phosphorylated substrate was eluted from the affinity membranes using a buffer containing 15% formic acid, and its absorbance was measured at 570 nm. PKC activity was estimated using a purified PKC from rat brain (0.02 units μl−1) as a standard and was measured as picomoles phosphate transferred per minute per miligram protein. Protein concentration was determined by the method of Bradford (Bradford, 1976) using bovine serum albumin (BSA) as a standard.
Chemicals and other reagents
Endothelin-1 (ET-1), PMA, bradykinin (BK), 5-HT, NG-nitro-L-arginine methylester (L-NAME) and sulphate salts of gentamicin, neomycin, kanamycin and streptomycin were obtained from Sigma. Sodium sulphate (15 mM) was used as a vehicle control for the aminoglycosides and was shown to be devoid of any effect on the muscle tension. Ro-32-0432 ([2-{8-[(Dimethylamino) methyl]-6,7,8,9-tetrahydropyridol[1,2-a]indol-3-yl}-3-(1-methylindol-3-y)maleimide, hydrochloride] was purchased from Calbiochem. PKC assay kit was obtained from Pierce. Bovine calf serum and Dulbecco's modified Eagle's medium (DMEM) were obtained from GIBCO Canada. All other reagents were of the highest available quality and were obtained from Sigma, Calbiochem or Fischer Scientific.
Statistical analysis of results
All results are reported as means±s.e.mean, with number of preparations used in parentheses. Statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Dunnet test when significant probability was reached. Values of P<0.05 were considered to be significant.
Results
ET-1 induced a concentration-dependent sustained contraction of cerebral arteries with an EC50 of 0.48 nM. This is in the range of ET-1 concentrations observed following SAH in patients (Seifert et al., 1995), and it may therefore be considered relevant in the pathophysiological setting. To elucidate the endothelin receptor subtype responsible for the ET-1-induced contraction, the responses to ET-1 were studied in cerebral arterial rings, in the presence or absence of a selective ETA receptor antagonist BQ-123, administered in the IC50 concentration (0.1 μM). As shown in Figure 1, exposure to this agent resulted in a competitive antagonism of the ET-1 responses.
Figure 1.
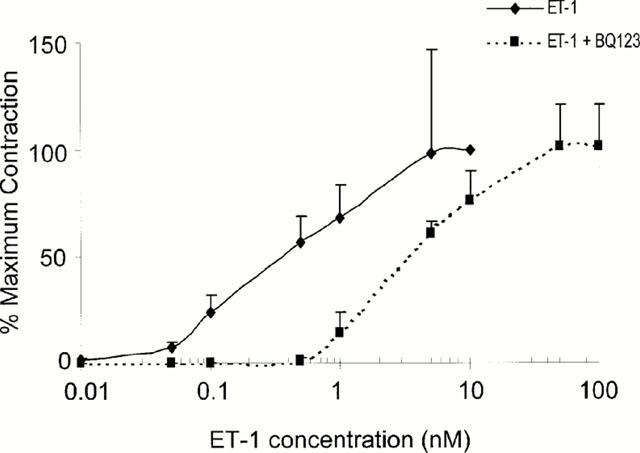
Concentration-effect curves for the vasoconstriction of rings of basilar artery elicited by endothelin-1 (0.5 nM) in the absence or presence of a selective antagonist of ETA receptor, BQ-123 (0.1 μM). Values are expressed as a percentage (% control) of the response observed in the absence of the antagonist. Data points represent means±s.e.mean (bars) for measurements with four to five individual ring preparations.
To study whether PKC was involved in the sustained phase of tonic contraction induced by ET-1, we examined the effects of a selective, cell permeable PKC inhibitor, Ro-32-0432, which displays 10 fold greater selectivity for the classical PKC isoforms (IC50 about 25 nM) over PKCε (IC50=180 nM) (Wilkinson et al., 1993). Ro-32-0432 (56 nM) administered to the preparations in which a plateau tension had developed in response to 0.5 nM ET-1, produced about 60% relaxation; a complete blockade was obtained at a 360 nM of the inhibitor (Figure 2). In contrast to ET-1 induced contraction, which consisted of both phasic and tonic components (Figure 3a), PMA caused only a sustained tonic contraction, slow in onset but progressive in the rise of tension, and it took about 50 – 60 min before a maximum tension developed (Figure 3b). Ro-32-0432 (56 nM) was less effective in the preparations precontracted with PMA (160 nM), and the maximal relaxation was obtained with a concentration of the inhibitor equal to 360 nM (Figure 2).
Figure 2.
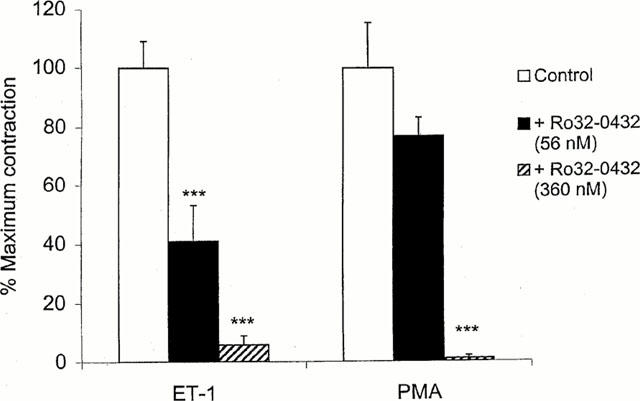
Effects of the PKC inhibitor, Ro-32-0432, on vasoconstriction induced in rings of basilar artery by endothelin-1 (ET-1) (0.5 nM) and PMA (160 nM). The ring preparations were first exposed to ET-1 or PMA, and Ro-32-0432 was administered after tonic response to the agents had developed. The responses are expressed as the percentage of maximum tonic tension observed in the absence of inhibitor (ordinate). Each bar represents mean±s.e.mean of 4 – 5 experiments. ***P<0.001, compared with control responses.
Figure 3.
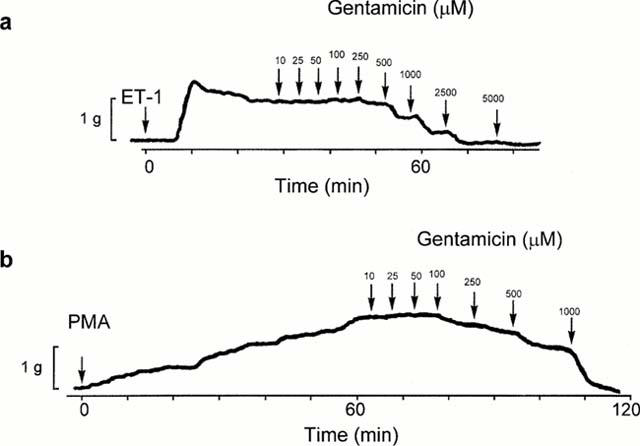
Representative traces of the effects of gentamicin on the responses of ring preparations pretreated with ET-1 (a) or PMA (b). Arrows indicate time of administration of ET-1, PMA and gentamicin. Each tracing illustrating the response of an individual preparation is representative of three to five independently conducted experiments.
In the study in which the effects of the aminoglycoside antibiotics were examined, neomycin, gentamicin, streptomycin and kanamycin were administered in increasing cumulative concentrations (0.01 – 10 mM) to ring preparations of canine basilar artery in which a tonic contraction to ET-1 (0.5 nM) or PMA (160 nM) had developed. Representative traces of the effects of gentamicin on the responses of tissues pretreated with ET-1 or PMA are shown in Figure 3. The relaxation of the preparations exposed to the aminoglycosides was expressed as the percentage of maximum tension produced by the vasoconstrictors. Cumulative concentration-effect curves for the aminoglycosides are shown in Figure 4a. The most effective agents were gentamicin and neomycin, both of which produced half-maximal relaxation of the contractions induced by ET-1 at 0.5 mM and 0.6 mM, respectively. Streptomycin and kanamycin were less effective and the EC50s for these agents were about 2 and 1.9 mM, respectively (Table 1). This order of potency reflects the number of positively charged amino groups; neomycin and gentamicin which possess six and five positively charged amino groups, respectively, were most effective against contractions caused by ET-1, while streptomycin and kanamycin characterized by the presence of three and four positive charges were less potent in producing relaxation. The relaxation was maintained over a prolonged time of exposure (several hours) to the aminoglycosides. In order to explore the possibility that vasodilatory effects of the aminoglycosides on ET-1 responses involve inhibition of PKC activity, we also studied the effects of these agents on the contractions elicited with PMA (160 nM).
Figure 4.
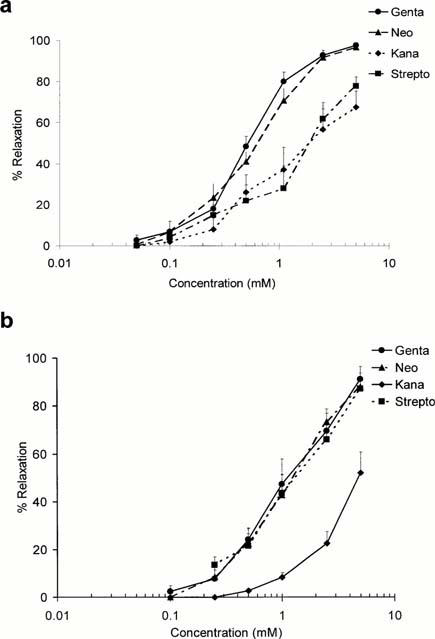
Cumulative concentration-effect curves for the relaxation of rings of canine basilar artery produced by the aminoglycosides. The preparations were first exposed to ET-1 (0.5 nM) (a) or PMA (160 nM) (b) and the aminoglycosides, gentamicin (genta), neomycin (neo), kanamycin (kana) and streptomycin (strepto), were administered in increasing cumulative concentrations to preparations in which maximum tonic tension had developed. Ordinate: relaxation expressed as a percentage of the maximum tension induced by ET-1. Abscissa: concentration of the aminoglycosides on a logarithmic scale. Data points represent the means±s.e.mean for experiments conducted with five or more ring preparations from three or more animals.
Table 1.
EC50 values (mM) for the aminoglycoside antibiotics against the vasoconstriction produced by endothelin-1 and phorbol 12-myristate 13 acetate (PMA)

Vasoconstriction induced by PMA started within 5 – 10 min, reached a maximum at about 60 min, and persisted for several hours. As shown in Figure 4b, neomycin, gentamicin and streptomycin induced about 80% of maximum relaxation of the contractions induced with PMA while kanamycin was less effective (about 52% of maximal relaxation). To determine whether inhibition of calcium influx could contribute to the effects of the aminoglycosides on contraction mediated by PMA, arterial preparations were exposed to these agents in calcium free medium. As shown in Figure 5, the PMA-induced contraction was maintained in calcium free buffer, and was attenuated by gentamicin (Figure 5a) and Ro-32-432, an inhibitor of PKC (Figure 5b), administered after a plateau tension had developed.
Figure 5.
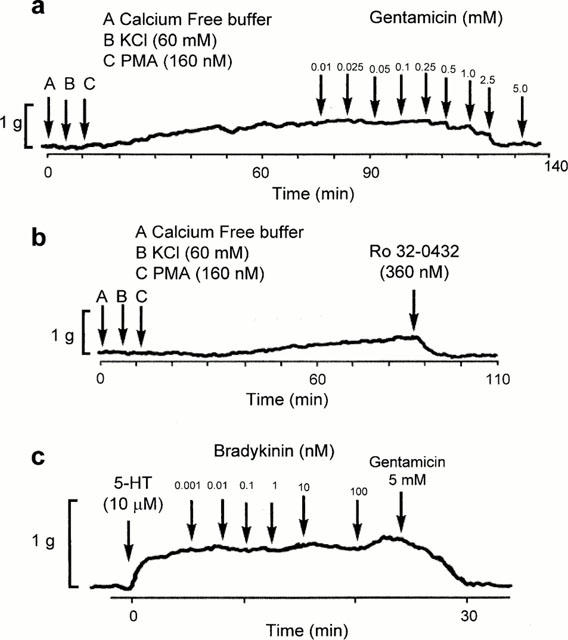
Representative traces of the vasoconstrictor responses induced by PMA (160 nM) in calcium free medium. Gentamicin (a) and Ro-32-0432 (b) were administered to the preparations in which maximum tonic contraction to PMA had developed. (c) shows representative traces of the effects of bradykinin on rings of basilar artery with denuded endothelium. The rings were precontracted with 5-HT (10 μM). Bradykinin produced no relaxation suggesting that endothelium had been removed from the preparations. Each trace, which illustrates the response of an individual ring preparation, is representative of three or four independent experiments.
In order to determine whether the vasorelaxant effects of the aminoglycosides could arise from an effect mediated by endothelium, responses to bradykinin were recorded as an indicator of the preservation of endothelial cells. Bradykinin (1 pM – 100 nM) produced a concentration-dependent vascular relaxation, in preparations which had been precontracted with 5-HT, in the presence but not in the absence of endothelium. In the arterial preparations in which bradykinin had no relaxant effect, gentamicin retained its relaxing activity thus indicating that the vasodilatory effect of this agent was endothelium independent (Figure 5c). In another series of experiments, rings were pre-incubated with an inhibitor of nitric oxide synthase, NG-nitro-L-arginine methylester (L-NAME) (100 μM), for 30 min. L-NAME did not significantly affect the contractions induced by ET-1, and relaxation caused by gentamicin was identical for rings incubated in the buffer with or without this inhibitor (results not shown).
Direct measurements of the total PKC activity were performed using cultured canine CVSMC stimulated with ET-1 (0.5 nM) and PMA (160 nM), in the presence or absence of the aminoglycosides. Since activated PKC translocates to the plasma and/or nucleus membranes, a process which is a hallmark of activation, the activity of this enzyme(s) was determined in both cytosolic and membrane (particulate) fractions. PKC activity was determined at the time-point (60 min) corresponding to maximum tonic contraction developed in response to ET-1 and PMA. The data show that a major effect of ET-1 and PMA is to elevate PKC activity in the particulate fractions of CVSMC exposed to these agents. There were corresponding decreases in the cytosolic fractions of cells treated with ET-1 and PMA, suggesting that challenge with these vasoconstrictors resulted in the PKC translocation. However, these changes did not reach statistical significance. The administration of gentamicin, neomycin, kanamycin and streptomycin, in the concentrations corresponding to the EC50 values determined in contractility studies, significantly reduced the effects of ET-1 on PKC activity in particulate fractions to 111±11.5%, 107±13%, 122±15% and 129±12% of control, respectively (Figure 6a). The effects of PMA on PKC activity were significantly reduced to 103±7%, 100±9%, 114±5.4% and 88.6±4.5% of control for gentamicin, neomycin, kanamycin and streptomycin, respectively (Figure 6b).
Figure 6.
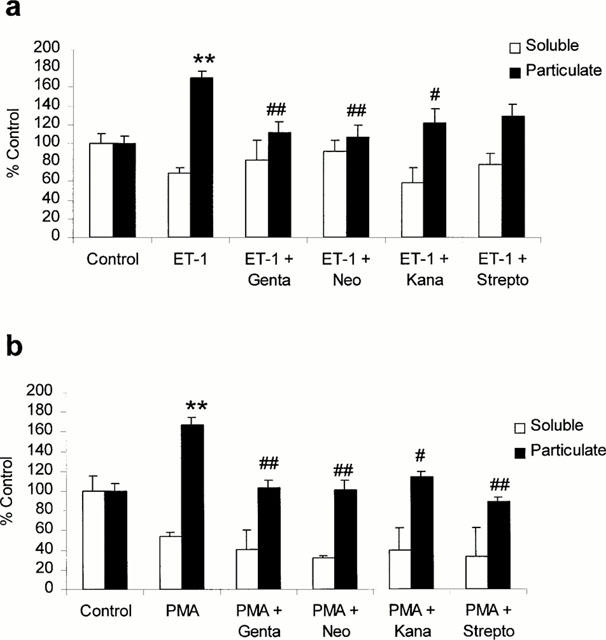
Effects of the aminoglycosides on protein kinase C (PKC) activity in cultured canine cerebrovascular smooth muscle cells. PKC activity was determined in the soluble and particulate fractions of serum starved and (48 h) cells exposed to ET-1 (0.5 nM) (a) or PMA (160 nM) (b) for 60 min. The aminoglycosides, gentamicin (genta), neomycin (neo), kanamycin (kana) and streptomycin (strepto), were administered 30 min after ET-1 application, at the concentrations corresponding to their EC50s determined in contractility experiments. Each bar represents mean value±s.e.mean of six experiments. **P<0.01 vs control; #P<0.01 vs stimulation with ET-1 alone; ##P<0.001 vs stimulation with ET-1 alone.
Discussion
The present studies demonstrate that PKC activation is a major component of a sustained vasoconstriction mediated by ET-1 in cerebral vessels. This is supported by the observations that Ro-32-0432, a selective inhibitor of the PKC isoforms (Wilkinson et al., 1993; Birchall et al., 1994), reversed vasoconstriction induced by ET-1 in canine basilar arteries. The inhibitory effects were evident with the concentrations of this agent capable to inhibit the classical and novel PKC enzymes, suggesting that both calcium-dependent and independent PKC's may be involved in the vasoconstrictor action of ET-1. Ro-32-0432 also reversed a tonic contraction produced by PMA in calcium free medium, indicating that the vasoconstrictor effects of PMA were mediated by a calcium independent PKC isoform. The contractile responses mediated by ET-1 were inhibited by the ETA receptors selective antagonist, BQ-123 in a competitive manner, indicating that the action of this peptide is likely to occur in cerebral arteries via the ET-1 receptor subtype coupled with PKC signaling pathway. The role of PKC in sustained vasoconstriction was further supported by direct studies of the PKC activity in CVSMC, which have shown that ET-1 stimulates this enzyme in CVSMC, at the time corresponding to maximum tonic contraction developed in cerebral arteries after exposure to this peptide and PMA.
It has been demonstrated that smooth muscle contraction is initiated by phosphorylation of myosin light chain (MLC), a process mediated by the calcium-calmodulin dependent myosin light chain kinase (MLCK) (Somlyo & Somlyo, 1994). Nevertheless, other data have shown that MLC phosphorylation does not participate in arterial narrowing during sustained vasospasm following SAH (Sun et al., 1998). These observations suggest that another mechanism may be important for the development of vasospasm, and there is evidence to suggest that this mechanism may involve the PKC mediated phosphorylation (Minami et al., 1992; Sako et al., 1993). How PKC might initiate vasospasm is unclear, but an important step may be the phosphorylation of the actin binding proteins, calponin and, indirectly, caldesmon, which inhibit myosin ATP-ase activity (Horowitz et al., 1996; Walsh et al., 1996). It has been reported that ET-1 stimulates phosphorylation of calponin mediated by PKC in peripheral blood vessels (Mino et al., 1995). Thus, it is conceivable that activation of PKC by this peptide in cerebral arteries and subsequent phosphorylation of actin thin filaments proteins play a role in the development of sustained vasoconstriction.
Another significant observation to emerge from the present studies is that gentamicin and the structurally related aminoglycosides are capable of attenuating of ET-1 effects in cerebral arteries and CVSMC. The vasodilatory effects were highly significant, endothelium independent, and maintained over a prolonged time of exposure to the aminoglycosides. The ability of the aminoglycosides to reverse the PMA-induced sustained contraction in the calcium-free medium, indicate that the effects of the aminoglycosides were indeed the consequence of PKC inhibition. Further support to this notion was provided by the observations that the aminoglycosides inhibited directly PKC activity in CVSMC exposed to ET-1 and PMA. Collectively these results suggest that the vasodilatory effects of the aminoglycosides arise from the inhibition of PKC activity in cerebrovascular smooth muscle. This suggestion is consistent with the observation that aminoglycosides prevent protein phosphorylation mediated by phorbol esters in cultured epithelial cells (Hagiwara et al., 1988).
The precise mechanism by which aminoglycosides inhibit PKC-mediated protein phosphorylation is unclear but it is likely that inhibition of this enzyme is due to the interaction of these compounds with acidic, negatively charged phospholipids in the plasma membrane. The aminoglycosides are polybasic due to their protonated amino groups and anionic phospholipids of the plasma membrane are prime targets for a charge interaction with these compounds (Lullmann & Vollmer, 1982). The observation that potencies of aminoglycoside antibiotics to inhibit PKC correspond to a number of ionizable amino groups of compounds support this contention (Hagiwara et al., 1988). It is therefore conceivable that the aminoglycosides could interfere with the association of protein kinase C with phosphatidylserine, a cofactor necessary for activity of this enzyme, and consequently modulate its ability to respond to activation by DAG and to phosphorylate target proteins. It is also likely that aminoglycosides could interfere with the conversion of phosphatidic acid, derived from phosphatidylcholine breakdown, to DAG, an effect believed to be responsible for sustained vasoconstriction (Asano & Matsui, 1999). Obviously, the elucidation of the molecular mechanism of PKC inhibition will require additional studies.
Although our data provide a strong argument in favour of a role of PKC in cerebral vasoconstriction, other possible modes of action for the vasodilatory effects of the aminoglycosides cannot be discounted. These agents exhibit various actions on several targets. Neomycin and the structurally related aminoglycosides inhibit inositol lipid-specific PLC and a variety of calcium channels including voltage-dependent L-type calcium channel (Langton et al., 1996), and calcium release-activated channels known to be stimulated by capacitative calcium entry (Kim et al., 1999). Furthermore, we have shown in our previous studies, that delayed administration of the aminoglycosides to the cerebrovascular cells stimulated with haemoglobin, can be very effective in decreasing the sustained elevation of intracellular calcium (Gergawy et al., 1998). Since ET-1 stimulates PLC activity and calcium influx mediated by a number of voltage dependent and independent calcium channels in smooth muscle cells (Nakajima et al., 1996; Iwamuro et al., 1999), it is likely that relaxation of the vasoconstriction induced with ET-1 may arise from multiple actions of the aminoglycosides.
The present findings support and extend our previous studies with aminoglycosides in several key ways. First, the findings provide evidence that the aminoglycosides are able to reverse the vasoconstrictor action of ET-1 with EC50s similar to those seen in our previous studies with haemoglobin and PGF2α (Gergawy et al., 1998). These effects were both significant and maintained over prolonged time. Second, our results demonstrate that delayed administration of the aminoglycosides to the cerebrovascular cells exposed to ET-1, can be effective in decreasing the sustained activation of PKC activity implicated in the development of vasospasm. The systemic administration of the aminoglycosides is not likely to be useful, because these agents do not cross the blood – brain barrier. However, intracranial application of a slow release formulation during surgical clipping of the aneurysm (Shiokawa et al., 1998) remains a realistic possibility.
Since we have shown in both our previous and the present studies that the aminoglycosides inhibited the vasoconstriction of multiple spasmogens implicated in the pathogenesis of cerebral vasospasm, a clear advantage may exist over specific antagonists or signal transduction inhibitors. It is now recognized that a number of pharmacologically unrelated factors are involved in the pathogenesis of cerebral vasospasm. The implication of such observations is that, for successful management of abnormal vasoconstriction, efficacious compounds may need to interfere with common signalling pathways shared by such factors. This carries with it the implication that aminoglycosides which inhibit several cellular actions, would be the promising agents in the search for pharmacological means of reversing the development of vasospasm and preventing cerebral ischaemia.
Acknowledgments
This work was supported by the Alberta Heart and Stroke Foundation.
Abbreviations
- CSF
cerebrospinal fluid
- CVSMC
cerebrovascular smooth muscle cells
- DAG
diacylglycerol
- ET
endothelin
- PKC
protein kinase C
- PMA
phorbol-12-myristate-13-acetate
- SAH
subarachnoid haemorrhage
References
- ASANO T, MATSUI T. Antioxidant therapy against cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Cell. Molec. Neurobiol. 1999;19:31–44. doi: 10.1023/a:1006908422937. [DOI] [PubMed] [Google Scholar]
- BIRCHALL A.M., BISHOP J., BRADSHAW D., CLINE A., COFFEY J., ELLIOTT L.M., GIBSON V.M., GREENHAM A., HALLAM T.J., HARRIS W., HILL C.H., HUTCHINGS A., LAMONT A.G., LAWTON G., LEWIS E.J., MAW A., NIXON J.S., POLE D., WADSWORTH J, WILKINSON S.E. Ro-32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J. Pharmacol. Exp. Ther. 1994;268:922–929. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- COOK D.A, VOLLRATH B. Free radicals and intracellular events associated with cerebrovascular spasm. Cardiovascular Res. 1995;30:493–500. [PubMed] [Google Scholar]
- ESKILDSEN-HELMOND Y.E., BEZSTAROSTI K., DEKKERS D.H., VAN HEUGTEN H.A, LAMERS J.M. Cross-talk between receptor-mediated phospholipase C-beta and D via protein kinase C as intracellular signal possibly leading to hypertrophy in serum free cultured cardiomyocytes. J. Molec. Cell. Cardiol. 1997;29:2545–2559. doi: 10.1006/jmcc.1997.0491. [DOI] [PubMed] [Google Scholar]
- GERGAWY M., VOLLRATH B, COOK D. The mechanism by which aminoglycoside antibiotics cause vasodilation of canine cerebral arteries. Br. J. Pharmacol. 1998;125:1150–1157. doi: 10.1038/sj.bjp.0702180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDIE R.G. Endothelins in health and disease: an overview. Clin. Exper. Pharmacol. Physiol. 1999;26:145–148. doi: 10.1046/j.1440-1681.1999.03014.x. [DOI] [PubMed] [Google Scholar]
- HAGIWARA M., INAGAKI M., KANAMURA K., OHTA H, HIDAKA H. Inhibitory effects of aminoglycosides on renal protein phosphorylation by protein kinase C. J. Pharmacol. Exp. Ther. 1988;244:355–360. [PubMed] [Google Scholar]
- HINO A., WEIR B.K.A., MACDONALD R.L., THISTED R., KIM C.J, JOHNS L.M. Prospective, randomized, double-blind trial of BQ-123 and bosentan for prevention of vasospasm following subarachnoid hemorrhage in monkeys. J. Neurosurg. 1995;83:503–509. doi: 10.3171/jns.1995.83.3.0503. [DOI] [PubMed] [Google Scholar]
- HOROWITZ A., MENICE C.B., LAPORTE R, MORGAN K.G. Mechanism of smooth muscle contraction. Physiol. Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- ITOH S., SASAKI T., ASAI A, KUCHINO Y. Prevention of delayed vasospasm by an endothelin ETA receptor antagonist, BQ-123: change of ETA receptor mRNA expression in a canine subarachnoid hemorrhage model. J. Neurosurg. 1994;81:759–764. doi: 10.3171/jns.1994.81.5.0759. [DOI] [PubMed] [Google Scholar]
- IWAMURO Y., MIWA S., ZHANG X.-F., MINOWA T., ENOKI T., OKAMOTO Y., HASEGAWA H., FURUTANI H., OKAZAWA M., ISHIKAWA M., HASHIMOTO N, MASAKI T. Activation of three types of voltage-independent Ca2+ channel in A7r5 cells by endothelin-1 as revealed by a novel Ca2+ channel blocker LOE 908. Br. J. Pharmacol. 1999;126:1107–1114. doi: 10.1038/sj.bjp.0702416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOSKO J., HENDRYK S., JEDRZEJOWSKA-SZYPULA H., GWOZDZ B., HERMAN Z.S, GAWLIK R. Influence endothelin ETA receptor antagonist BQ-123 on changes of endothelin-1 level in plasma of rats with acute vasospasm following subarachnoid hemorrhage. J. Physiol. Pharmacol. 1998;49:367–375. [PubMed] [Google Scholar]
- KASUYA H., WEIR B.K.A., WHITE D.M, STEFANSON K. Mechanism of oxyhemoglobin-induced release of endothelin-1 from cultured vascular endothelial cells and smooth muscle cells. J. Neurosurg. 1993;79:892–898. doi: 10.3171/jns.1993.79.6.0892. [DOI] [PubMed] [Google Scholar]
- KIM K.T., CHOI S.Y, PARK T.J. Neomycin inhibits catecholamine secretion by blocking nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. J. Pharmacol. Exp. Ther. 1999;288:73–80. [PubMed] [Google Scholar]
- LANGTON P.D., FARLEY R, EVERITT D.E. Neomycin inhibits K+-induced force and Ca2+ channel current in rat arterial smooth muscle. Pflügers Arch. - Eur. J. Physiol. 1996;433:188–193. doi: 10.1007/s004240050266. [DOI] [PubMed] [Google Scholar]
- LULLMANN H, VOLLMER B. An interaction of aminoglycoside antibiotics with Ca2+ binding to lipid monolayers and to biomembranes. Biochem. Pharmacol. 1982;31:3769–3773. doi: 10.1016/0006-2952(82)90291-x. [DOI] [PubMed] [Google Scholar]
- MINAMI N., TANI E., MAEDA Y., YAMAURA I, FUKAMI M. Effects of inhibitors of protein kinase C and calpain in experimental delayed cerebral vasospasm. J. Neurosurg. 1992;76:111–118. doi: 10.3171/jns.1992.76.1.0111. [DOI] [PubMed] [Google Scholar]
- MINO T., YUASA U., NAKA M, TANAKA T. Phosphorylation of calponin mediated by protein kinase C in association with contraction in porcine coronary artery. Biochem. Biophys. Res. Commun. 1995;208:397–404. doi: 10.1006/bbrc.1995.1351. [DOI] [PubMed] [Google Scholar]
- NAKAJIMA T., HAZAMA H., HAMADA E., WU S.-N., IGARASHI K., YAMASHITA T., SEYAMA Y., OMATA M, KURACHI Y. Endothelin-1 and vasopressin activate Ca2+-permeable non-selective cation channels in aortic smooth muscle cells: Mechanism of receptor-mediated Ca2+ influx. J. Mol. Cell. Cardiol. 1996;28:707–722. doi: 10.1006/jmcc.1996.0066. [DOI] [PubMed] [Google Scholar]
- OHLSTEIN E.H, STORER B.L. Oxyhemoglobin stimulation of endothelin production in cultured endothelial cells. J. Neurosurg. 1992;77:274–278. doi: 10.3171/jns.1992.77.2.0274. [DOI] [PubMed] [Google Scholar]
- PERRIER M.L., SCATTON B, BENAVIDES J. Dihydropyridine and omega-conotoxin resistant, neomycin sensitive calcium channels mediate the depolarization induced increase in internal calcium levels in cortical slices from immature rat brain. J. Pharmacol. Exp. Ther. 1992;261:324–330. [PubMed] [Google Scholar]
- PICHLER M., WANG Z., GRABNER-WEISS C., REIMER D., HERING S, GRABNER M. Block of P/Q-type calcium channels by therapeutic concentrations of aminoglycoside antibiotics. Biochemistry. 1996;35:14659–14664. doi: 10.1021/bi961657t. [DOI] [PubMed] [Google Scholar]
- RUMENAPP U., SCHMIDT M., WAHN F., TAPP E., GRANNASS A, JAKOBS K.H. Characteristics of protein kinase C- and ADP-ribosylation factor-stimulated phospholipase D activities in human embryonic kidney cells. Eur. J. Biochem. 1997;248:407–414. doi: 10.1111/j.1432-1033.1997.00407.x. [DOI] [PubMed] [Google Scholar]
- SAKO M., NISHIHARO J, OHTA S. Role of protein kinase C in the pathogenesis of cerebral vasospasm after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 1993;13:247–254. doi: 10.1038/jcbfm.1993.30. [DOI] [PubMed] [Google Scholar]
- SEIFERT V., LOFFLER B.M., ZIMMERMANN M., ROUX S, STOLKE D. Endothelin concentration in patients with aneurysmal subarachnoid hemorrhage: Correlation with cerebral vasospasm, delayed ischemic neurological deficits and volume of hematoma. J. Neurosurg. 1995;82:55–62. doi: 10.3171/jns.1995.82.1.0055. [DOI] [PubMed] [Google Scholar]
- SCHIFFRIN E.L, TOUYZ R.M. Vascular biology of endothelin. J. Cardiovasc. Pharmacol. 1998;32 Suppl 3:S2–S13. [PubMed] [Google Scholar]
- SCHINI V.B., HENDRICKSON H, HEUBLEIN D.M. Thrombin enhances the release of endothelin from porcine aortic endothelial cells. Eur. J. Pharmacol. 1989;165:333–334. doi: 10.1016/0014-2999(89)90733-4. [DOI] [PubMed] [Google Scholar]
- SHIOKAWA K., KASUYA H., MIYAIMA M., IZAWA M, TAKAKURA K. Prophylactic effect of papaverine prolonged release pellets on cerebral vasospasm in dogs. Neurosurgery. 1998;42:109–115. doi: 10.1097/00006123-199801000-00022. [DOI] [PubMed] [Google Scholar]
- SOMLYO A.P, SOMLYO A.V. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- SUN H., KANAMARU K., MASAAKI I., SUZUKI H., KOJIMA T., WAGA S., KUREISHI Y, NAKANO T. Myosin light chain phosphorylation and contractile proteins in a canine two-hemorrhage model of subarachnoid hemorrhage. Stroke. 1998;29:2149–2154. doi: 10.1161/01.str.29.10.2149. [DOI] [PubMed] [Google Scholar]
- WALSH M.P., HOROWITZ A., CLEMENT-CHOMIENNE O., ANDREA J.E., ALLEN B.G, MORGAN K.G. Protein kinase C mediation of Ca2+-independent contractions of vascular smooth muscle. Biochem. Cell Biol. 1996;74:485–502. doi: 10.1139/o96-053. [DOI] [PubMed] [Google Scholar]
- VOLLRATH B., CHAN P., FINDLAY M, COOK D. Lazaroids and deferoxamine attenuate the intracellular effects of oxyhemoglobin in vascular smooth muscle. Cardiovasc. Res. 1995;30:619–626. [PubMed] [Google Scholar]
- VOLLRATH B.A.M., WEIR B.K.A., MACDONALD R.L, COOK D.A. Intracellular mechanisms involved in the responses of cerebrovascular smooth muscle cells to hemoglobin. J. Neurosurg. 1994;80:261–268. doi: 10.3171/jns.1994.80.2.0261. [DOI] [PubMed] [Google Scholar]
- WANEBO J.E., ARTHUR A.S., LOUIS H.G., WEST K., KASSELL N.F., LEE K.S, HELM G.A. Systemic administration of the endothelin-A receptor antagonist TBC 11251 attenuates cerebral vasospasm after experimental subarachnoid hemorrhage: dose study and review of endothelin-based therapies in the literature on cerebral vasospasm. Neurosurgery. 1998;43:1409–1417. [PubMed] [Google Scholar]
- WILKINSON S.E., PARKER P.J, NIXON J.S. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J. 1993;294:335–337. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIMMERMANN M, SEIFERT V. Endothelin and subarachnoid hemorrhage: An overview. Neurosurgery. 1998;43:863–876. doi: 10.1097/00006123-199810000-00083. [DOI] [PubMed] [Google Scholar]