

Abstract
The effects of tachykinin NK1, NK2 and NK3 receptor agonists and antagonists were measured on blood pressure (MAP) and heart rate (HR) after bilateral microinjection into the substantia nigra (SN) of awake, unrestrained rats.
Increasing doses (25 pmol – 1 nmol) of selective agonists at NK1 ([Sar9,Met(O2)11]SP), NK2 ([β-Ala8]NKA(4 – 10)) and NK3 (senktide) receptors into the SN produced tachycardia which was selectively and reversibly blocked by the prior injection of tachykinin antagonists at NK1 (RP67580, 250 pmol), NK2 (SR48968, 250 pmol) and NK3 (R-820, 500 pmol) receptor. A rapid fall in MAP followed by a pressor response was seen with 1 nmol of [Sar9,Met(O2)11]SP. Behavioural activity was elicited by 1 nmol of [Sar9,Met(O211]SP (sniffing>face washing=grooming) and senktide (sniffing>wet dog shake>rearing=locomotion). Tachykinin antagonists had no direct cardiovascular or behavioural effects.
The tachycardia produced by 100 pmol of [β-Ala8]NKA(4 – 10) or senktide was abolished by an i.v. treatment with atenolol (β1-adrenoceptor antagonist, 5 mg kg−1) while that evoked by [Sar9,Met(O2)11]SP was reduced. A combination of atenolol (5 mg kg−1) and atropine (muscarinic antagonist, 1 mg kg−1) blocked the response evoked by [Sar9,Met(O2)11]SP.
These data suggest that the SN is a potential site of modulation of cardiac activity by tachykinins. In addition to the withdrawal of the cardiovagal activity by NK1 receptor, the three tachykinin receptors appear to increase the sympatho/adrenal drive to the heart. This occurs independently of changes in MAP and behaviour. Hence, this study highlights a new central regulatory mechanism of cardiac autonomic activity.
Keywords: Tachykinin, tachykinin NK1, NK2 and NK3 receptors, substantia nigra, autonomic regulation, substance P
Introduction
The substantia nigra (SN ; A9) is a midbrain nucleus traditionally associated with the central control of motor activity (Gerfen, 1992). Additional evidence also suggests its involvement in central cardiovascular regulation (Barbeau et al., 1969; Micieli et al., 1987; Van den Buuse et al., 1991; Lin & Yang, 1994). Electrical or chemical stimulation of SN leads to an increase in mean arterial pressure (MAP), heart rate (HR) and striatal dopamine levels in anaesthetized rats (Lin & Yang, 1994). Since these cardiovascular effects were markedly attenuated by nigral 6-hydroxydopamine lesions or by blockade of striatal dopamine receptors, it was suggested that the nigro-striatal dopaminergic pathway is involved in these responses (Lin & Yang, 1994). Lesion of the nigro-striatal dopaminergic pathway attenuates the development of hypertension in young spontaneously hypertensive rats that points to a putative role for this neuronal pathway in the development of hypertension (Van den Buuse et al., 1991; Linthorst et al., 1994).
Tachykinins are known to modulate nigro-striatal dopaminergic neurons by stimulating the release, turnover and metabolism of striatal dopamine via the activation of neurokinin-1 (NK1) and neurokinin-3 (NK3) receptors in the SN (Reid et al., 1990; Humpel et al., 1991; Humpel & Saria, 1993; Bannon & Whitty, 1995; Marco et al., 1998). Autoradiographic, immunohistochemical and in situ and/or solution hybridization studies revealed that the SN contains numerous substance P (SP) and neurokinin A (NKA) containing nerve terminals and tachykinin NK1 and NK2 receptors (Helke et al., 1990; Stoessl & Hill, 1990; Stoessl, 1994; Bannon & Whitty, 1995; Whitty et al., 1995; Shughrue et al., 1996; Futami et al., 1998). Also, SP and NKA are released in vivo (Lindefors et al., 1989) and in vitro (Jessell, 1978; Humpel & Saria, 1989) in the SN from striato-nigral neurons which are thought to function as a positive feedback loop which regulates the activity of the nigro-striatal dopaminergic pathway.
Previous work from our laboratory has shown that central activation of tachykinin NK3 receptors by intracerebroventricular (i.c.v.) injection of the selective agonist senktide causes increases of mean arterial pressure, heart rate and behavioural activity in conscious rats (Cellier et al., 1997). Intravenous pre-treatment with the dopamine D2 receptor antagonist haloperidol blocked those cardiovascular and behavioural changes and unmasked a vasopressin-dependent bradycardia (Cellier & Couture, 1997). Although anatomical and physiological studies suggest an interaction between tachykinins and the nigro-striatal dopamine pathway in the SN, there is a lack of information regarding the implication of tachykinins in cardiovascular regulation at the level of the SN. Therefore, the present study was undertaken to test the hypothesis that tachykinin receptors may modulate cardiovascular function following the activation of the nigro-striatal dopaminergic pathway. This was addressed by investigating the cardiovascular responses to bilateral microinjection of selective tachykinin agonists and antagonists at NK1 ([Sar9,Met(O2)11]SP, Regoli et al., 1988; RP 67580, Garret et al., 1991), NK2 ([β-Ala8]NKA(4 – 10), Rovero et al., 1989; SR 48968, Advenier et al., 1992; Emonds-Alt et al., 1992) and NK3 (senktide, Wörmser et al., 1986; R-820, Regoli et al., 1994) receptors directly into the SN. The pseudopeptide NK3 antagonist R-820 (Regoli et al., 1994) was preferred to the non-peptide NK3 antagonist SR 142801 (Emonds-Alt et al., 1995) in this study because the latter was shown to act as a tachykinin NK3 receptor agonist when injected intracerebroventricularly or intrathecally in conscious rats (Cellier et al., 1997; Couture et al., 2000). The mechanism underlying the cardiovascular response to agonists was investigated by the prior intravenous injection of the selective β1-adrenoceptor antagonist atenolol and the muscarinic antagonist atropine methyl nitrate. To avoid the spurious effects of anaesthesia and the stress induced by immobilization, these studies were carried out in awake, unrestrained rats.
Methods
Animal source and care
Male Wistar rats (300 – 400 g) were purchased 3 – 5 days prior to experiments from Charles River, St-Constant, Québec, Canada and housed four to five per cage under a 12 h light – dark cycle in a room with controlled temperature (20°C), humidity (53%) with food (Charles River Rodent) and tap water available ad libitum. The care of animals and research protocols conformed to the guiding principles for animal experimentation as enunciated by the Canadian Council on Animal Care and approved by the Animal Care Committee of our University.
Animal preparation
Male Wistar rats (n=154) were anaesthetized with an intraperitoneal (i.p.) injection of 65 mg kg−1 sodium pentobarbitone (Somnotol; M.T.C. Pharmaceuticals, Cambridge, Ontario, Canada) and then positioned in a stereotaxic frame (David Kopf Instrumentation, Tujunga, CA, U.S.A.) with the incisor bar set at 3.3 mm below the interaural line. The skull was exposed, cleaned and a hole was drilled above each SN (coordinates : 5.3 mm posterior to the bregma, 2.3 mm lateral to the midline, 7.0 mm ventral from the skull surface ; Paxinos & Watson, 1986). Two 23-gauge stainless steel guide cannulae targeted 2 mm dorsal to each SN were implanted and fixed with two screws and dental cement to the skull. Stylets (31-gauge stainless steel) were inserted into each guide cannulae. Then, the skin was replaced and sutured. Animals were housed in individual plastic cages (40×23×20 cm) in the same controlled conditions and allowed to recover for 7 days. Then, the rats were re-anaesthetized with sodium pentobarbitone (65 mg kg−1, i.p.) and an intravascular siliconized (Sigmacote, Sigma, St-Louis, MO, U.S.A.) polyethylene tubing PE-50 catheter (Intramedics, Clay Adams, NJ, U.S.A.), filled with physiological saline containing 100 i.u. ml−1 heparin sodium salt (Sigma, St-Louis, MO, U.S.A.), was inserted into the abdominal aorta via the femoral artery for direct blood pressure recording. The catheter was tunnelled subcutaneously to emerge at the back of the neck. Before surgery, the animals received Ethacilin (5 mg kg−1, i.m., rogar/S.T.B. Inc., London, Ontario, Canada) and Ketoprophen (anafen, 10 mg kg−1, i.m., MERIAL Canada Inc., Baie d'Urfé, Québec, Canada). Recovery from anaesthesia was monitored closely under a warming lamp to maintain the body temperature of animals. Rats with apparent abnormal behaviour (loss of >25% of body weight, anorexia, weaknesses) were immediately humanely killed with an overdose of pentobarbitone. Thereafter, rats were housed individually in polyethylene cages with a top grid and returned to their resident room. Experimental protocols were initiated 48 h after the final intervention, in conscious and unrestrained rats.
Measurement of cardiovascular and behavioural parameters
During all experiments, continuous direct recordings of blood pressure and heart rate were made respectively with a Statham pressure Transducer (P23ID) and a cardiac tachometer (model 7P4) (triggered by the arterial blood pressure pulse) coupled to a Grass polygraph (model 79; Grass Instruments Co., Quincy, MA, U.S.A.). Behavioural activity was measured as previously reported (Picard et al., 1994). Briefly, during every consecutive period of 15 s, a score of 1 or 0 was given systematically depending on whether the animal showed the specific type of behaviour or not, whatever its frequency, intensity or duration during that period. Summation of scores for the first 30 min period following SN injection gave the behavioural scores for face washing, grooming, sniffing and rearing. The maximal theoretical score was 120 (15 s intervals×30 min). Wet dog shakes and locomotion (number of complete exploratory circles within the cage) behaviours were measured according to the number of episodes or frequency during the first 30 min period. Both cardiovascular and behavioural responses were measured 1 h after the rats were transported to an isolated and quiet testing room where only the experimenter had access. Rats remained in their resident cage but the top grid was removed and they had no more access to the food and water for the duration of the experiment. When resting blood pressure and heart rate were stable, bilateral microinjections were made simultaneously into each SN of undisturbed, freely moving rats through 31-gauge stainless steel injectors that extend 2 mm beyond the previously implanted guide cannulae. The injectors were connected, via polyethylene tubing (PE-10, Intramedics, Clay Adams, NJ, U.S.A.), to two Hamilton microsyringes (5 μl, Fisher Scientific Ltd, Montréal, Québec, Canada) and inserted into the guide cannulae without handling the rats. All solutions for microinjections were freshly prepared and injected (volume of 0.1 μl) bilaterally into the SN over a period of 1 min.
Histology
At the end of the experiments, the rats received 0.1 μl of Evans Blue dye (Sigma, St Louis, MO, U.S.A.) bilaterally and they were immediately sacrificed with an overdose of sodium pentobarbitone. The brains were removed and fixed with 10% (v v−1) formol and 20% (w v−1) sucrose. Coronal sections (40 μm, cut on a freezing microtome) were mounted on glass slides and stained with cresyl violet for histological examination of the microinjection sites (Figure 1). Rats which showed any evidence of haemorrhage (n=39) were excluded from the study.
Figure 1.
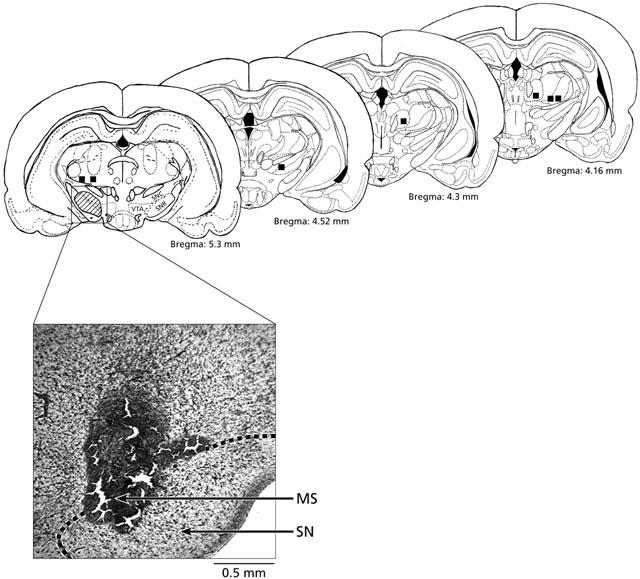
Identification of the microinjection sites in the substantia nigra (SN) (shaded areas) following post-mortem histological examination of microinjected Evans's blue (0.1 μl bilaterally). The hatched zone represents accepted microinjection sites and the squares represent microinjection sites (n=7) excluded from the results and kept as negative control for spread. A rat was considered successfully injected when both cannula tips were shown to be slightly above SN or within a distance of 0.5 mm of the SN (AP=5.3 mm posterior to the bregma) (Paxinos & Watson, 1986). Abbreviations: MS, microinjection site; SN, substantia nigra; SNC, SN pars compacta; SNR, SN pars reticulata; VTA, ventral tegmental area. Scale: 0.5 mm.
Experimental protocols
Experiment 1: dose-response curves to SN microinjection of selective tachykinin agonists
The rats were used on 4 consecutive days, during which they received initially a SN microinjection of artificial cerebrospinal fluid (aCSF) followed 60 min later by the injection of the selective NK1 agonist [Sar9,Met(O2)11]SP (n=18), the selective NK2 agonist [β-Ala8]NKA(4 – 10) (n=13) and the selective NK3 agonist senktide (n=19) at random. Increasing doses of agonists were given at 24 h apart on day 1 (25 pmol), day 2 (50 pmol), day 3 (100 pmol) and day 4 (1 nmol, the largest dose). On the same day, rats received a single dose of the three agonists at a minimum of 60 min intervals to enable blood pressure and heart rate to return to baseline values. Because the HR response to agonists at 1 nmol was prolonged and returned only slowly to pre-injection levels, only one or two agonists were given to a rat at this dose. Every dose refers to the summation of doses given on each side of the SN.
Preliminary studies were performed to control for tachyphylaxis. For that purpose, rats were used on 2 consecutive days during which they received one dose of a maximum of two different agonists at NK1 (25 pmol, n=8 and 1 nmol, n=6), NK2 (25 pmol, n=9 and 1 nmol, n=3) and NK3 receptor (1 nmol, n=3). Results showed a reproducible response to agonists when injected either at 25 pmol or 1 nmol 1 day apart. In addition, dose-response curves on heart rate response (intensity and time-course) to NK1, NK2 and NK3 agonists were not significantly different (ANOVA, two-way) whatever the order of injection of agonists on day 1 to 4, suggesting that there is no cross desensitization between the three agonists.
Experiment 2: cardiovascular effects of SN microinjection of selective tachykinin antagonists
Rats that had previously (24 h) received 25 pmol of either [Sar9,Met(O2)11]SP, [β-Ala8]NKA (4 – 10) or 50 pmol of senktide (the lowest dose which produced significant tachycardia) were given an intra-nigral microinjection of the NK1 antagonist RP67580 (250 pmol against [Sar9,Met(O2)11]SP (n=10) or [β-Ala8]NKA (4 – 10) (n=4)), the NK2 antagonist SR48968 (250 pmol against [β-Ala8]NKA (4 – 10) (n=6 – 8) or [Sar9,Met(O2)11]SP (n=6)) and the NK3 antagonist R-820 (500 pmol against senktide (n=7 – 8) or [Sar9,Met(O2)11]SP (n=8)); after 15 min, intra-nigral microinjections of 25 or 50 pmol of the respective agonists were given. At least 120 min later, the same antagonist was re-injected and after 15 min, an agonist for a different receptor was given to test the selectivity of the antagonist. Each rat received only one antagonist. The agonists were re-injected alone 24 h later to assess the reversibility of any inhibition occurring on the preceding day in the presence of antagonist. Doses of antagonists were based on pilot studies in which several doses of antagonists were tested to identify that required to block the cardiovascular effect of agonists. Preliminary studies revealed that an equimolar dose (1 nmol) of the NK3 antagonist, R-820, did not alter the cardiovascular and behavioural effects of the NK3 agonist, senktide (n=5). The doses of 1 and 10 nmol of R-820 were excluded because they required, respectively 30% (v v−1) and 60% (v v−1) of dimethyl sulphoxide (DMSO) which were not tolerated by the animals. Therefore, this study was restricted to the dose of 500 pmol of R-820 with 15% (v v−1) DMSO which did not produce side effects.
Experiment 3: mechanism of the cardiovascular responses to tachykinin agonists
Rats that had previously (24 h) received 100 pmol of either [Sar9,Met(O2)11]SP, [β-Ala8]NKA (4 – 10) or senktide were given an i.v. injection of the selective β1-adrenoceptor antagonist atenolol (5 mg kg−1) or a combination of atenolol (5 mg kg−1) and the cholinergic muscarinic antagonist atropine methyl nitrate (1 mg kg−1) simultaneously. Doses of the latter antagonists were based on a previous study (Décarie & Couture, 1992). Intra-nigral microinjection of 100 pmol of [Sar9,Met(O2)11]SP (n=7), [β-Ala8]NKA (4 – 10) (n=14) and senktide (n=7) were given 10 min after the treatment. On the same day, each rat received a maximum of three microinjections into the SN at a minimum of 60 min intervals. The treatment with atropine alone was not performed because in pilot experiments we found that heart rate was markedly increased under cholinergic muscarinic receptor blockade. As a consequence, the ceiling effect of atropine masked the tachycardia produced by the tachykinin agonists injected into the SN. However, MAP was not affected by atropine given alone.
Drugs and solutions
The composition of aCSF was, in mM: NaCl 128.6, KCl 2.6, MgCl2 2.0 and CaCl2 1.4; pH adjusted to 7.2. [Sar9,Met(O2)11]SP was obtained from Peninsula Lab. Inc. (San Carlos, CA, U.S.A.) while [β-Ala8]NKA (4 – 10) and succinyl-[Asp6, MePhe8]SP-(6 – 11) (senktide) were purchased from Bachem Bioscience Inc. (King of Prussia, PA, U.S.A.). The antagonists RP67580 and SR 48968 were provided by Dr C. Garret (Rhone Poulenc, Paris, France) and J.-C. Brelière (Sanofi Recherche, Montpellier, France), respectively. The pseudopeptide antagonist R-820 was generously provided by Dr J.L. Fauchère (Servier, Paris, France). Atenolol and atropine methyl nitrate were purchased from Sigma Chemicals Co (St Louis, MO, U.S.A.) and solubilized in saline (0.9% w v−1) for i.v. injection. [Sar9,Met(O2)11]SP was solubilized in aCSF while senktide, [β-Ala8]NKA (4 – 10) and all antagonists were solubilized in DMSO (Fisher Scientific, Montréal, Québec, Canada). The solution was then completed with aCSF and contained 1 – 15% (v v−1) DMSO. The stock solutions (10 mg ml−1) of agonists and antagonists were stored in aliquots of 100 μl at −20°C until use. In all experiments, vehicle (aCSF containing <15% DMSO) was injected as control and produced no significant changes on any parameters when compared to baseline values.
Statistical analysis of data
Results are expressed as means±s.e.mean of (n) rats. Results were analysed for statistical significance by a two-way analysis of variance (ANOVA) with repeated measures followed by Bonferroni confidence intervals. Statistical differences on non-parametric episodes of behaviours were evaluated with a Kruskal-Wallis test. Only probability values (P) less than 0.05 were considered to be statistically significant.
Results
Cardiovascular response induced by the NK1 agonist [Sar9,Met(O2)11]SP
The effects of four increasing doses of [Sar9,Met(O2)11]SP on MAP and HR are shown in Figure 2. [Sar9,Met(O2)11]SP (25 pmol – 1 nmol) evoked dose-dependent increases in HR which were significant (P<0.05) when compared to vehicle (aCSF) values (n=18). Thus, the tachycardia was significant at 25 pmol (0.5 – 15 min, n=17), 50 pmol (0.5 – 15 min, n=18), 100 pmol (0.5 – 45 min, n=12) and 1 nmol (0.5 – 60 min, n=8). The maximum rise in HR was 44±7 b.p.m. (25 pmol, 15 min), 31±11 b.p.m. (50 pmol, 15 min), 75±18 b.p.m. (100 pmol, 15 min) and 105±25 b.p.m. (1 nmol, 3 min). Whereas [Sar9,Met(O2)11]SP had no effect on MAP at doses between 25 – 100 pmol, it produced a rapid and transient fall in MAP (−12±2 mmHg) at 1 nmol (0.5 – 1 min, P<0.05, n=8). The above NK1 agonist (1 nmol) produced a small but significant increase in MAP (8±3 mmHg) (P<0.05) at 45 – 60 min post-injection.
Figure 2.
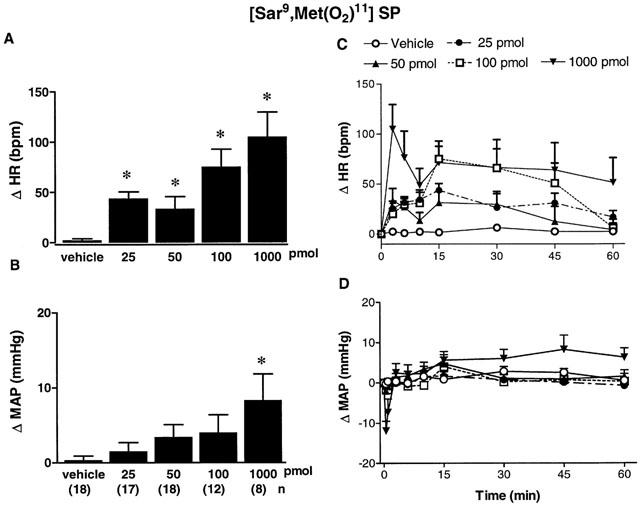
Maximal changes (A,B) and time-course effects (C,D) on heart rate (Δ HR, A,C) and mean arterial pressure (Δ MAP, B,D) following bilateral SN microinjection of vehicle (aCSF) or increasing doses of the NK1 agonist [Sar9,Met(O2)11]SP from 25 pmol to 1 nmol in conscious rats. Each point represents the mean±s.e.mean of (n) rats. Comparison to vehicle values is indicated by *P<0.05 (one-way ANOVA followed by a test of Bonferroni). Basal values were: MAP: 105±8 mmHg and HR:347±12 b.p.m.
Cardiovascular response induced by the NK2 agonist [β-Ala8]NKA (4 – 10)
The effects of four increasing doses of [β-Ala8]NKA (4 – 10) on MAP and HR are shown in Figure 3. [β-Ala8]NKA (4 – 10) evoked a tachycardia which was significant (P<0.05) at 25 pmol (0.5 – 30 min, n=13), 50 pmol (0.5 – 45 min, n=14), 100 pmol (2 – 45 min, n=12) and 1 nmol (0.5 – 45 min, n=18) when compared to vehicle (aCSF with 5 – 10% DMSO) values (n=14). The maximum rise in HR was 41±9 b.p.m. (25 pmol, 15 min), 48±15 b.p.m. (50 pmol, 15 min), 48±15 b.p.m. (100 pmol, 15 min) and 36±16 b.p.m. (1 nmol, 30 min). [β-Ala8]NKA (4 – 10) (25 – 1000 pmol) had no significant effect on MAP when compared to vehicle values.
Figure 3.
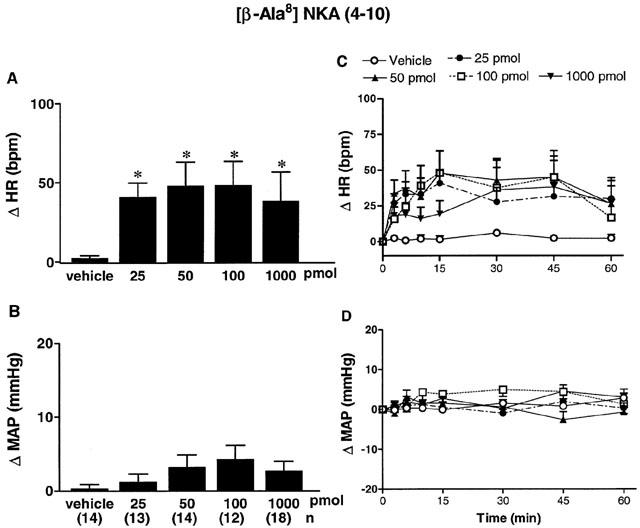
Maximal changes (A,B) and time-course effects (C,D) on heart rate (Δ HR, A,C) and mean arterial pressure (Δ MAP, B,D) following bilateral SN microinjection of vehicle (aCSF with 5 – 10% DMSO) or increasing doses of the NK2 agonist [β-Ala8]NKA (4 – 10) from 25 pmol to 1 nmol in conscious rats. Each point represents the mean±s.e.mean of (n) rats. Comparison to vehicle values is indicated by *P<0.05 (one-way ANOVA followed by a test of Bonferroni). Basal values were: MAP: 108±4 mmHg and HR: 339±13 b.p.m.
Cardiovascular response induced by the NK3 agonist senktide
The effects of four increasing doses of senktide on MAP and HR are shown in Figure 4. Senktide evoked a tachycardia which was significant (P<0.05) at 50 pmol (0.5 – 15 min, n=18), 100 pmol (0.5 – 60 min, n=13) and 1 nmol (0.5 – 45 min, n=9). The maximum rise in HR was 26±7 b.p.m. (25 pmol, 7 min, P>0.05), 42±12 (50 pmol, 15 min), 69±14 (100 pmol, 15 min) and 47±16 (1 nmol, 45 min). However, senktide had no significant effect on MAP when compared to vehicle values (aCSF with 10% DMSO) at any doses tested.
Figure 4.
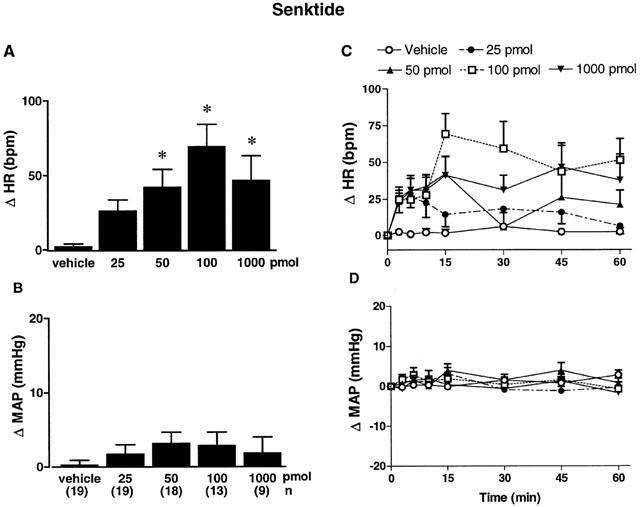
Maximal changes (A,B) and time-course effects (C,D) on heart rate (Δ HR, A,C) and mean arterial pressure (Δ MAP, B,D) following bilateral SN microinjection of vehicle (aCSF with 10% DMSO) or increasing doses of the NK3 agonist senktide from 25 pmol to 1 nmol in conscious, unrestrained rats. Each point represents the mean±s.e.mean of (n) rats. Comparison to vehicle values is indicated by *P<0.05 (one-way ANOVA followed by a test of Bonferroni). Basal values were: MAP: 107±4 mmHg and HR: 343±14 b.p.m.
Behavioural effects of [Sar9,Met(O2)11]SP, [β-Ala8]NKA (4 – 10) and senktide
No significant behavioural activity was associated with the injection of the three agonists from 25 to 100 pmol. At 1 nmol, [β-Ala8]NKA (4 – 10) (n=18) did not produce any significant effect on behaviour while [Sar9,Met(O2)11]SP evoked a significant increase (P<0.01, n=8) of behavioural activity (sniffing>wet dog shake>face washing=grooming=head scratching). Whereas senktide (1 nmol) produced a significant (P<0.01, n=9) increase on some of these behaviours (sniffing>wet dog shake>rearing=locomotion), it did not affect face washing, head scratching and grooming (Figure 5). The behaviours produced by the NK1 and NK3 receptor agonists occurred concomitantly with the cardiovascular responses and presented a similar time course.
Figure 5.
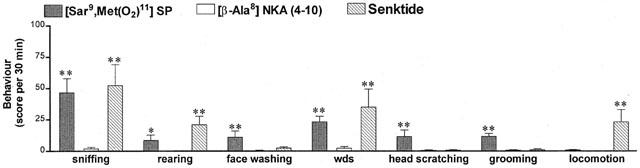
Bar graphs showing maximum behavioural response (score per 30 min) including sniffing, rearing, face washing, wet dog shakes (wds), head scratching, grooming and locomotion following bilateral SN microinjection of 1 nmol of [Sar9,Met(O2)11]SP (n=8), [β-Ala8]NKA (4 – 10) (n=18) and senktide (n=13). Each bar represents the mean±s.e.mean of (n) rats. *P<0.05 and **P<0.01 when compared to vehicle values (n=18, not shown as close to zero values) with a Kruskal-Wallis test.
Effects of RP67580 on [Sar9,Met(O2)11]SP- and [β-Ala8]NKA (4 – 10)-induced responses
The effects of the tachykinin NK1 antagonist RP67580 against the tachycardiac responses to [Sar9,Met(O2)11]SP and [β-Ala8]NKA (4 – 10) are shown in Figure 6A. On day 1, the responses to [Sar9,Met(O2)11]SP (25 pmol, n=10) and [β-Ala8]NKA (4 – 10) (25 pmol, n=4) were quantitatively similar to those obtained in Figures 2 and 3. The peak response was reached at 10 min after injection of [Sar9,Met(O2)11]SP (51±20 b.p.m.) and 3 min after injection of [β-Ala8]NKA (4 – 10) (30±5 b.p.m.). On day 2, the response to [Sar9,Met(O2)11]SP was abolished (P<0.05) in the presence of RP67580 (250 pmol, n=10) and was restored 24 h later (44±24 b.p.m.). The same treatment with this antagonist had no significant effect on the response to [β-Ala8]NKA (4 – 10) (43±24 b.p.m.) (Figure 6A).
Figure 6.
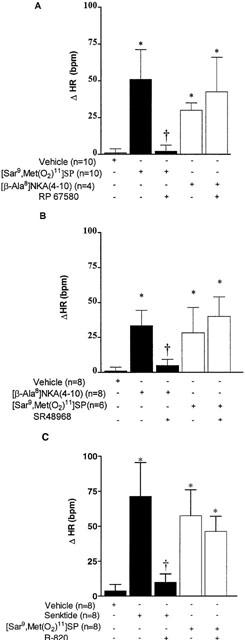
Bar graphs showing maximum heart rate responses (ΔHR) following bilateral SN microinjection of [Sar9,Met(O2)11]SP (25 pmol in A and B or 50 pmol in C), [β-Ala8]NKA (4 – 10) (25 pmol) and senktide (50 pmol) in the absence (−) and presence (+) of (A) RP67580 (250 pmol), (B) SR 48968 (250 pmol) and (C) R-820 (500 pmol) in conscious rats. Each point represents the mean±s.e.mean of (n) rats. Statistical comparison was made to vehicle values (*P<0.05) or to the agonist alone (†P<0.05), using one-way ANOVA followed by a test of Bonferroni.
Effects of SR48968 on [β-Ala8]NKA (4 – 10)- and [Sar9,Met(O2)11]SP-induced responses
The effects of the tachykinin NK2 antagonist SR48968 against the tachycardiac responses to [β-Ala8]NKA (4 – 10) and [Sar9,Met(O2)11]SP are shown in Figure 6B. On day 1, the responses to [β-Ala8]NKA (4 – 10) (25 pmol, n=8) and [Sar9,Met(O2)11]SP (25 pmol, n=6) were quantitatively similar to those presented in Figures 2 and 3. The response to [Sar9,Met(O2)11]SP peaked at 15 min after injection (28±18 b.p.m.) while that to [β-Ala8]NKA (4 – 10) peaked at 2 min after injection (33±11 b.p.m.). On day 2, the response to [β-Ala8]NKA (4 – 10) was blocked (P<0.05) in the presence of SR48968 (250 pmol, n=8) and was restored 24 h after treatment (47±15 b.p.m.). However, SR48968 failed to alter significantly the response to [Sar9,Met(O2)11]SP (40±14 b.p.m.).
Effects of R-820 on senktide- and [Sar9,Met(O2)11]SP-induced responses
The effects of the tachykinin NK3 antagonist R-820 against the tachycardiac responses to senktide and [Sar9,Met(O2)11]SP are shown in Figure 6C. On day 1, the responses to senktide (50 pmol, n=8) and [Sar9,Met(O2)11]SP (50 pmol, n=8) were quantitatively similar to those presented in Figures 2 and 4. The response peaked at 30 min after injection of senktide (71±24 b.p.m.) and 15 min after injection of [Sar9, Met(O2)11]SP (58±19 b.p.m.). On day 2, the response to senktide was blocked (P<0.05) by R-820 (500 pmol, n=8) and was restored 24 h after treatment (43±24 b.p.m.). However, R-820 failed to alter significantly the response to [Sar9,Met(O2)11]SP (46±11 b.p.m.).
When injected alone, the three antagonists (250 pmol of RP67580 and SR48968, 500 pmol of R-820) failed to elicit any significant cardiovascular or behavioural effects over a period of 60 min compared to vehicle values (data not shown).
Effects of atenolol and atropine on [Sar9,Met(O2)11]SP-induced responses
The tachycardia induced by [Sar9,Met(O2)11]SP in the presence of atenolol and atropine are shown in Figure 7A. On day 1, in non-atenolol pretreated rats, the response to [Sar9,Met(O2)11]SP (100 pmol, n=7) was significant between 1 – 45 min. The response peaked at 15 min after the injection (54±14 b.p.m.). On day 2, the response induced by [Sar9,Met(O2)11]SP was not significantly reduced by the prior injection of atenolol (5 mg kg−1, i.v.) although the remaining tachycardia was no more significant when compared to vehicle values. In a second group of rats, in non-atenolol and non-atropine pretreated rats, [Sar9,Met(O2)11]SP (100 pmol, n=7) evoked a similar tachycardia on day 1. On day 2, the response induced by [Sar9,Met(O2)11]SP was completely blocked (P<0.05) by the prior simultaneous injection of atenolol (5 mg kg−1, i.v.) and atropine methyl nitrate (1 mg kg−1, i.v.).
Figure 7.
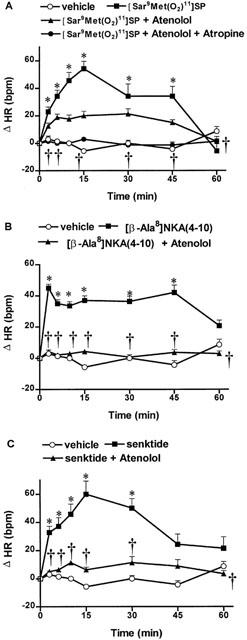
Time-course effects on changes in heart rate (ΔHR) following bilateral SN microinjection of vehicle (aCSF +10% DMSO, n=7 – 14), A [Sar9,Met(O2)11]SP (100 pmol, n=7), B [β-Ala8]NKA (4 – 10) (100 pmol, n=14) and C senktide (100 pmol, n=7) in the absence or presence of atenolol (5 mg kg−1, i.v.) or a combination of atenolol (5 mg kg−1, i.v.) and atropine (1 mg kg−1, i.v.) in conscious rats. Each point represents the mean±s.e.mean of (n) rats. Statistical comparison was made to vehicle values (*P<0.05) or to the agonist alone (†P<0.05) (two-way ANOVA followed by a test of Bonferroni).
Effect of atenolol on [β-Ala8]NKA (4 – 10)-induced responses
The tachycardia induced by [β-Ala8]NKA (4 – 10) in the presence of atenolol is shown in Figure 7B. On day 1, in non-atenolol pretreated rats, the response to [β-Ala8]NKA (4 – 10) (100 pmol, n=14) was quantitatively similar (significant 1 – 45 min) to that presented in Figure 3. The response peaked at 2 min after the injection (45±9 b.p.m.). On day 2, the response induced by [β-Ala8]NKA (4 – 10) was completely blocked (P<0.05) by the prior injection of atenolol (5 mg kg−1, i.v.).
Effect of atenolol on senktide-induced responses
The tachycardia induced by senktide in the presence of atenolol is shown in Figure 7C. On day 1, in non-atenolol pretreated rats, the response to senktide (100 pmol, n=7) was quantitatively similar (significant 4 – 30 min) to that presented in Figure 4. The response peaked at 15 min after the injection (60±24 b.p.m.). On day 2, the response induced by senktide was completely blocked (P<0.05) by the prior injection of atenolol (5 mg kg−1, i.v.).
Effects of agonists in control experiments
Rats which showed injection sites beside the SN (black squares, Figure 1; after histological post-mortem examination) failed to evoke any significant cardiovascular and behavioural changes to [Sar9,Met(O2)11]SP (100 pmol, n=7), [β-Ala8]NKA (4 – 10) (100 pmol, n=4) or senktide (100 pmol, n=4) compared to vehicle values (data not shown). Therefore, the effects of agonists are unlikely to be due to the diffusion of the injection outside the SN. This possibility is also unlikely in this study because the volume (0.1 μl) of injection is 5 to 10 fold smaller than those (0.5 and 1 μl) generally employed in this nucleus (Humpel et al., 1991; Humpel & Saria, 1993; Stoessl et al., 1993; 1995). Finally, among the rats in which microinjection sites were accepted in this study, no differences could be seen in the size of the heart rate response whether the injection was within or very close to the pars compacta or pars reticulata of the SN.
Discussion
Several lines of evidence support the role of tachykinins in central cardiovascular regulation (Culman & Unger, 1995; Culman et al., 1997; Picard et al., 1994; Cellier et al., 1997; 1999) and in the modulation of the nigro-striatal dopaminergic system (Reid et al., 1990; Humpel et al., 1991; Humpel & Saria, 1993; Marco et al., 1998). However, this study provides the first pharmacological evidence suggesting a putative role of these striato-nigral neuropeptides in cardiovascular regulation at the level of the substantia nigra.
Bilateral microinjections of rather low doses (pmol range) of selective tachykinin NK1, NK2 and NK3 receptor agonists into SN produced significant tachycardia in awake, unrestrained rats. These effects are unlikely to be due to locomotor activity or to blood pressure changes because alterations in MAP (NK1 agonist) and behaviour (NK1 and NK3 agonists) occurred only at the highest dose of agonists (1 nmol). Thus, these data confirm earlier studies showing that tachykinins enhance behavioural activity at the nmol range doses by activating NK1 and NK3 receptors in the SN (Stoessl et al., 1993; 1995). Blood pressure was not significantly altered with tackykinin agonists except for the highest dose (1 nmol) of the NK1 agonist. Indeed, a rapid and transient fall in MAP was observed followed by a late and small increase in MAP. This early fall in MAP may have activated the baroreflex and thus overestimated the tachycardiac response observed at this dose. In contrast to blood pressure and behavioural activity, low doses (25 – 50 pmol) of the tachykinin agonists at NK1, NK2 and NK3 receptors were sufficient to modify heart rate. This observation suggests a physiological role of these neuropeptides on heart rate modulation by a direct action in the SN. The tachycardia induced by the selective agonists was further characterized with highly selective antagonists and supports the involvement of the three tachykinin receptors. The antagonists tested blocked in a selective and reversible manner the heart rate increase produced by each agonist. Our hypothesis involving the three receptors is reinforced by the fact that the time-course of the cardiac response is different for each agonist. For instance, the tachycardia elicited by senktide was prolonged, in contrast to the rapid and short-lasting tachycardia produced by [Sar9, Met(O2)11]SP. Also, the heart rate response to the NK2 agonist was not dose-dependent. This may be due to the relatively low density of NK2 receptors in SN unlike the abundance of NK1 and especially NK3 receptor binding sites, protein and mRNAs (Stoessl & Hill, 1990; Bannon & Whitty, 1995; Whitty et al., 1995; Shughrue et al., 1996; Futami et al., 1998). Although a barely detectable signal of NK2 receptor mRNA was seen in this nucleus (Bannon & Whitty, 1995; Whitty et al., 1995), we showed a weak immunoreactivity for the NK2 receptor in some fibres and cell bodies of medium size in the rostral SN pars reticulata (Zerari et al., 1996).
At the doses tested, the three antagonists did not produce any significant behavioural effect. This is consistent with a previous study showing that NK1 (RP67580), NK2 (L-659,877) and NK3 ([Trp7,β-Ala8]NKA (4 – 10)) receptor antagonists (2 nmol) had no effect on grooming behaviour per se (Stoessl et al., 1995). Tachykinin antagonists were also devoid of any cardiovascular activity. This suggests that endogenous tachykinins may not play a primary role in the tonic control of blood pressure and heart rate at the level of the SN in normal rats. This is consistent with the general concept that tachykinins are not tonically active but are released in response to noxious stimuli and stress (Unger et al., 1988; Culman & Unger, 1995; Culman et al., 1997; Baulmann et al., 2000). Nevertheless, the high sensitivity of the heart rate response to exogenous tachykinins suggests a potential role of these neuropeptides in the control of baroreflex activity in the SN.
Treatment with atropine methyl nitrate alone was not performed in this study because its i.v. injection caused a large tachycardia which masked that evoked by tachykinin agonists in the SN. Blockade of ganglionic nicotinic receptors by atropine is unlikely as this treatment did not affect blood pressure. The tachycardia which occurs in the presence of atropine is most likely the consequence of the withdrawal of the vagal tone to the heart. In the presence of atenolol, the tachycardiac responses to the NK2 and NK3 agonists were abolished, suggesting the involvement of a sympathetic mechanism mediated by β1-adrenoceptors. In contrast, the tachycardia induced by the NK1 agonist was completely blocked only by a combination of atenolol and atropine, suggesting a double contribution of cardiac sympathetic fibres and a reduction of the cardiovagal drive in the response. It is therefore feasible that the NK1 receptor stimulates a neural pathway different from that activated by NK2 and NK3 receptors. The identification of these putative pathways involved in the regulation of cardiac autonomic activity by intranigral tachykinins will deserve further investigations.
Moreover, the behavioural responses elicited by high doses of the NK1 and NK3 agonists in the SN had two different profiles. Whereas senktide enhanced sniffing, locomotion, rearing and wet dog shakes, [Sar9,Met(O2)11]SP increased face washing, grooming, sniffing, wet dog shakes and head scratching. In agreement with this finding, senktide (1 nmol) was reported to increase locomotion and wet dog shakes (Stoessl et al., 1993) while the NK1 agonist [AcArg6,Sar9,Met(O2)11]SP (6 – 11) (0.5 and 1 nmol) induced grooming (Stoessl et al., 1995). In the same matter, the distinctive effects of NK1 and NK3 receptor agonists on behaviour could be related to the activation of different neuronal elements in the SN. Indeed, in situ and/or solution hybridization, autoradiographic and immunocytochemical studies have shown that NK3 receptors are located mainly on dopaminergic neurons in the substantia nigra pars compacta (Stoessl & Hill, 1990; Stoessl, 1994; Bannon & Whitty, 1995; Whitty et al., 1995; Shughrue et al., 1996; Chen et al., 1998) while NK1 receptors are mostly located on GABAergic neurons of the substantia nigra pars reticulata (Sivam & Krause, 1992; Stoessl, 1994; Bannon & Whitty, 1995). Furthermore, an in vitro electrophysiological study revealed that the NK1 agonist, [Sar9,Met(O2)11]SP, activates non-dopaminergic neurons (presumably GABAergic which represent about 9% of neurons) in the guinea-pig SN pars compacta; in contrast, the NK3 agonist, senktide, activates dopaminergic neurons (the large majority with 78% of neurons) of the same area. In the latter study, the selectivity of the response to agonists was further confirmed with specific antagonists (Nalivaiko et al., 1997). An in vivo microdialysis approach in guinea-pig has shown that senktide injected in the SN pars compacta evokes an increase of extracellular dopamine concentration in the striatum which was blocked by a selective NK3 antagonist (Marco et al., 1998). Thus, the heart rate response induced by the NK1 agonist [Sar9,Met(O2)11]SP could result from the activation of a GABAergic pathway, possibly the nigro-thalamo-cortico-striatal loop while that induced by the NK3 agonist senktide may result from the direct activation of the nigro-striatal dopaminergic pathway. Further studies are required to confirm this hypothesis.
In conclusion, by using highly selective tachykinin receptor agonists and antagonists, it was shown that the activation of NK1, NK2 and NK3 receptors in the SN leads to consistent tachycardia by a mechanism independent of behavioural activity and changes in blood pressure. It is suggested that the activation of the NK2 and NK3 receptors into the SN evoked a tachycardia by enhancing the sympatho/adrenal drive to the heart while the NK1 agonist-induced tachycardia results from a reduction of the vagal tone in coordination with an increase of sympatho-adrenal activity. Thus, this study provides the first pharmacological evidence that tachykinin receptors in the substantia nigra are potentially involved in the central autonomic modulation of cardiac activity.
Acknowledgments
Authors acknowledge Dr C. Garret (Rhone Poulenc, France), Dr J. C. Brelière (Sanofi Recherche, France) and Dr J. L. Fauchère (Institut de recherches Servier, France) for the donation of RP67580, SR48968 and R-820, respectively. A. Lessard holds research traineeships from the Heart and Stroke Foundation of Canada and FRSQ-FCAR program. This work was supported by a grant from the Canadian Institutes of Health Research (MT-14379).
Abbreviations
- aCSF
artificial cerebrospinal fluid
- ANOVA
analysis of variance
- HR
heart rate
- i.v.
intravenous
- i.c.v.
intracerebroventricular
- MAP
mean arterial blood pressure
- NKA
neurokinin A
- SN
substantia nigra
- SHR
spontaneously hypertensive rat
- SP
substance P
References
- ADVENIER C., ROUISSI N., NGUYEN Q.T., EMONDS-ALT X., BRELIERE J.C., NELIAT G., NALINE E., REGOLI D. Neurokinin A (NK-2) receptor revisited with SR 48968, a potent non peptide antagonist. Biochem. Biophys. Res. Commun. 1992;184:1418–1424. doi: 10.1016/s0006-291x(05)80041-5. [DOI] [PubMed] [Google Scholar]
- BANNON M.J., WHITTY C.J. Neurokinin receptor gene expression in substantia nigra:localization, regulation, and potential physiological significance. Can. J. Physiol. Pharmacol. 1995;73:866–870. doi: 10.1139/y95-119. [DOI] [PubMed] [Google Scholar]
- BARBEAU A., GILLO-JOFFROY L., BOUCHER R., NOWACZYNSKI W., GENEST J. Renin-aldosterone system in Parkinson's disease. Science. 1969;165:291–292. doi: 10.1126/science.165.3890.291. [DOI] [PubMed] [Google Scholar]
- BAULMANN J., SPITZNAGEL H., HERDEGEN T., UNGER Th., CULMAN J. Tachykinin receptor inhibition and c-fos expression in the rat brain following formalin-induced pain. Neuroscience. 2000;95:813–820. doi: 10.1016/s0306-4522(99)00478-9. [DOI] [PubMed] [Google Scholar]
- CELLIER E., BARBOT L., IYENGAR S., COUTURE R. Characterization of central and peripheral effects of septide with the use of five tachykinin NK1 receptor antagonists in the rat. Br. J. Pharmacol. 1999;127:717–728. doi: 10.1038/sj.bjp.0702620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CELLIER E., BARBOT L., REGOLI D., COUTURE R. Cardiovascular and behavioural effects of intracerebroventricularly administered tachykinin NK3 receptor antagonists in the conscious rat. Br. J. Pharmacol. 1997;122:643–654. doi: 10.1038/sj.bjp.0701435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CELLIER E., COUTURE R.Mechanism of cardiovascular and behavioural responses to central NK-3 receptor activation in conscious rat 199740In International Tachykinin Conference: Tachykinins in Health and Disease, Cairns (Australia), September 7–11, Abstract book p
- CHEN L.-W., GUAN Z.-L., DING Y.-Q. Mesencephalic dopaminergic neurons expressing neuromedin K receptor (NK3): a double immunocytochemical study in the rat. Brain Res. 1998;780:150–154. [PubMed] [Google Scholar]
- COUTURE R., TOMA N., BARBOT L. SR142801 behaves as a tachykinin NK-3 receptor agonist on a spinal nociceptive reflex in the rat. Life Sci. 2000;66:51–65. doi: 10.1016/s0024-3205(99)00561-5. [DOI] [PubMed] [Google Scholar]
- CULMAN J., UNGER Th. Central tachykinins: mediators of defence reaction and stress reactions. Can. J. Physiol. Pharmacol. 1995;73:885–891. doi: 10.1139/y95-122. [DOI] [PubMed] [Google Scholar]
- CULMAN J., KLEE S., OHLENDORF C., UNGER Th. Effect of tachykinin receptor inhibition in the brain on cardiovascular and behavioral responses to stress. J. Pharmacol. Exp. Ther. 1997;280:238–246. [PubMed] [Google Scholar]
- DÉCARIE A., COUTURE R. Characterization of the peripheral action of neuropeptide K on the rat cardiovascular system. Eur. J. Pharmacol. 1992;213:125–131. doi: 10.1016/0014-2999(92)90241-u. [DOI] [PubMed] [Google Scholar]
- EMONDS-ALT X., BICHON D., DUCOUX J.P., HEAULME M., MILOUX B., PONCELET M., PROIETTO V., VAN BROECK D., VILAIN P., NELIAT G., SOUBRIE P., LE FUR G., BRELIERE J.C. SR 142801, the first potent non-peptide antagonist of the tachykinin NK3 receptor. Life Sci. 1995;56:27–32. doi: 10.1016/0024-3205(94)00413-m. [DOI] [PubMed] [Google Scholar]
- EMONDS-ALT X., VILAIN P., GOULAOUIC P., PROIETTO V., VAN BROECK D., ADVENIER C., NALINE E., NELIAT G., LEFUR G., BRELIERE J.C. A potent and selective non peptide antagonist of the neurokinin A (NK-2) receptor. Life Sci. 1992;50:PL101–PL106. doi: 10.1016/0024-3205(92)90352-p. [DOI] [PubMed] [Google Scholar]
- FUTAMI T., HATANAKA Y., MATSUSHITA K., FURUYA S. Expression of substance P receptor in the substantia nigra. Mol. Brain Res. 1998;54:183–198. doi: 10.1016/s0169-328x(97)00307-0. [DOI] [PubMed] [Google Scholar]
- GARRET C., CARRUETTE A., FARDIN V., MOUSSAOUI S., PEYRONEL J.F., BLANCHARD J.C., LADURON P.M. Pharmacological properties of a potent and selective nonpeptide substance P antagonist. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10208–10212. doi: 10.1073/pnas.88.22.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERFEN C.R. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Ann. Rev. Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- HELKE C.J., KRAUSE J.E., MANTYH P.W., COUTURE R., BANNON M.J. Diversity in mammalian tachykinin peptidergic neurons: multiple peptides, receptors, and regulatory mechanisms. FASEB J. 1990;4:1606–1615. [PubMed] [Google Scholar]
- HUMPEL C., SARIA A. Effects of GABA and L-glutamic acid on the potassium-evoked in vitro release of substance P and neurokinin A-like immunoreactivities are different in the rat striatum and substantia nigra. Neurosci. Lett. 1989;105:159–163. doi: 10.1016/0304-3940(89)90029-3. [DOI] [PubMed] [Google Scholar]
- HUMPEL C., SARIA A. Intranigral injection of selective neurokinin-1 and neurokinin-3 but not neurokinin-2 receptor agonists biphasically modulate striatal dopamine metabolism but not striatal preprotachykinin-A mRNA in the rat. Neurosci. Lett. 1993;157:223–226. doi: 10.1016/0304-3940(93)90742-4. [DOI] [PubMed] [Google Scholar]
- HUMPEL C., SARIA A., REGOLI D. Injection of tachykinins and selective neurokinin receptor ligands into the substantia nigra reticulata increases striatal dopamine and 5-hydroxytryptamine metabolism. Eur. J. Pharmacol. 1991;195:107–114. doi: 10.1016/0014-2999(91)90387-6. [DOI] [PubMed] [Google Scholar]
- JESSELL T.M. Substance P release from the rat substantia nigra. Brain Res. 1978;151:469–478. doi: 10.1016/0006-8993(78)91080-6. [DOI] [PubMed] [Google Scholar]
- LIN M.-T., YANG J.-J. Stimulation of the nigrostriatal dopamine system produces hypertension and tachycardia in rats. Am. J. Physiol. (Heart Circ. Physiol. 35) 1994;266:H2489–H2496. doi: 10.1152/ajpheart.1994.266.6.H2489. [DOI] [PubMed] [Google Scholar]
- LINDEFORS N., BRODIN E., TOSSMAN U., SEGOVIA J., UNGERSTEDT U. Tissue levels and in vivo release of tachykinins and GABA in striatum and substantia nigra of rat brain after unilateral striatal dopamine denervation. Expl. Brain Res. 1989;74:527–534. doi: 10.1007/BF00247354. [DOI] [PubMed] [Google Scholar]
- LINTHORST A.C.E., VAN GIERSBERGEN P.L.M., GRAS M., VERSTEEG D.H.G., DE JONG W. The nigrostriatal dopamine system : role in the development of hypertension in spontaneously hypertensive rats. Brain Res. 1994;639:261–268. doi: 10.1016/0006-8993(94)91739-6. [DOI] [PubMed] [Google Scholar]
- MARCO N., THIRION A., MONS G., BOUGAULT I., LE FUR G., SOUBRIÉ P., STEINBERG R. Activation of dopaminergic and cholinergic neurotransmission by tachykinin NK3 receptor stimulation : an in vivo microdialysis approach in guinea pig. Neuropeptides. 1998;32:481–488. doi: 10.1016/s0143-4179(98)90075-0. [DOI] [PubMed] [Google Scholar]
- MICIELI G., MARTIGNONI E., CAVALLINI A., SANDRINI G., NAPPI G. Postprandial and orthostatic hypotension in Parkinson's disease. Neurology. 1987;37:386–393. doi: 10.1212/wnl.37.3.386. [DOI] [PubMed] [Google Scholar]
- NALIVAIKO E., MICHAUD J.-C., SOUBRIÉ P., LE FUR G., FELTZ P. Tachykinin neurokinin-1 and neurokinin-3 receptor-mediated responses in guinea-pig substantia nigra:an in vitro electrophysiological study. Neuroscience. 1997;78:745–757. doi: 10.1016/s0306-4522(96)00625-2. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The rat brain in stereotaxic coordinates. Academic Press, Toronto, Canada, Harbourt Brace Jovanovich; 1986. [Google Scholar]
- PICARD P., REGOLI D., COUTURE R. Cardiovascular and behavioural effects of centrally administered tachykinins in the rat : characterization of receptors with selective antagonists. Br. J. Pharmacol. 1994;112:240–249. doi: 10.1111/j.1476-5381.1994.tb13058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REGOLI D., DRAPEAU G., DION S., COUTURE R. New selective agonists for neurokinin receptors : pharmacological tools for receptor characterization. Trends Pharmacol. Sci. 1988;9:290–295. doi: 10.1016/0165-6147(88)90013-2. [DOI] [PubMed] [Google Scholar]
- REGOLI D., NGUYEN Q.T., JUKIC D. Neurokinin receptor subtypes characterized by biological assays. Life Sci. 1994;54:2035–2047. doi: 10.1016/0024-3205(94)00712-8. [DOI] [PubMed] [Google Scholar]
- REID M.S., HERRERA-MARSCHITZ M., HÖKFELT T., LINDEFORS N., PERSSON H., UNGERSTEDT U. Striatonigral GABA, dynorphin, substance P and neurokinin A modulation of nigrostriatal dopamine release : evidence for direct regulatory mechanisms. Exp. Brain Res. 1990;82:293–303. doi: 10.1007/BF00231249. [DOI] [PubMed] [Google Scholar]
- ROVERO P., PESTELLINI V., PATACCHINI R., GIULIANI S., SANTICIOLI P., MAGGI C.A., MELI A., GIACHETTI A. A potent and selective agonist for the NK-2 tachykinin receptor. Peptides. 1989;10:593–595. doi: 10.1016/0196-9781(89)90148-4. [DOI] [PubMed] [Google Scholar]
- SHUGHRUE P.J., LANE M.V., MERCHENTHALER I. In situ hybridization analysis of the distribution of neurokinin-3 mRNA in the rat central nervous system. J. Comp. Neurol. 1996;372:395–414. doi: 10.1002/(SICI)1096-9861(19960826)372:3<395::AID-CNE5>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- SIVAM S.P., KRAUSE J.E. Tachykinin systems in the spinal cord and basal ganglia: influence of neonatal capsaicin treatment or dopaminergic intervention on levels of peptides, substance P-encoding mRNAs, and substance P receptor mRNA. J. Neurochem. 1992;59:2278–2284. doi: 10.1111/j.1471-4159.1992.tb10121.x. [DOI] [PubMed] [Google Scholar]
- STOESSL A.J. Localization of striatal and nigral tachykinin receptors in the rat. Brain Res. 1994;646:13–18. doi: 10.1016/0006-8993(94)90052-3. [DOI] [PubMed] [Google Scholar]
- STOESSL A.J., HILL D.R. Autoradiographic visualization of NK-3 tachykinin binding sites in the rat brain, utilizing [3H]senktide. Brain Res. 1990;534:1–7. doi: 10.1016/0006-8993(90)90105-k. [DOI] [PubMed] [Google Scholar]
- STOESSL A.J., BRACKSTONE M., RAJAKUMAR N., GIBSON C.J. Pharmacological characterization of grooming induced by a selective NK-1 tachykinin receptor agonist. Brain Res. 1995;700:115–120. doi: 10.1016/0006-8993(95)00940-r. [DOI] [PubMed] [Google Scholar]
- STOESSL A.J., POLANSKI E., FRYDRYSZAK H. Effects of ageing on tachykinin function in the basal ganglia. Brain Res. 1993;632:21–28. doi: 10.1016/0006-8993(93)91133-d. [DOI] [PubMed] [Google Scholar]
- UNGER Th., CAROLUS S., DEMMERT G., GANTEN D., LANG R.E., MASERGLUTH C., STEINBERG H., VEELKEN R. Substance P induces a cardiovascular defense reaction in the rat : pharmacological characterization. Circ. Res. 1988;63:812–820. doi: 10.1161/01.res.63.4.812. [DOI] [PubMed] [Google Scholar]
- VAN DEN BUUSE M., LINTHORST A.C.E., VERSTEEG D.H.G., DE JONG W. Role of brain dopamine systems in the development of hypertension in the spontaneously hypertensive rat. Clin. Exp. Hyp. 1991;A13:653–659. [Google Scholar]
- WHITTY C.J., WALKER P.D., GOEBEL D.J., POOSCH M.S., BANNON M.J. Quantitation, cellular localization and regulation of neurokinin receptor gene expression within the rat substantia nigra. Neuroscience. 1995;64:419–425. doi: 10.1016/0306-4522(94)00373-d. [DOI] [PubMed] [Google Scholar]
- WÖRMSER U., LAUFER R., HART Y., CHOREV M., GILON C., SELINGER Z. Highly selective agonists for substance P receptor subtypes. EMBO J. 1986;5:2805–2808. doi: 10.1002/j.1460-2075.1986.tb04571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZERARI F., DION S., KARPITSKIY V., KRAUSE J., COUTURE R.Immunocytochemical localization of tachykinin NK-2 receptors (NK-2R) in the rat central nervous system (CNS) 199639In international multidisciplinary symposium : Peptide Receptors, Montreal (Canada), July 28-August 1, Abstract book p