

Abstract
The anticancer anthracycline doxorubicin (DOX) causes cardiotoxicity. Enzymatic reduction of a side chain carbonyl group converts DOX to a secondary alcohol metabolite that has been implicated in cardiotoxicity. We therefore monitored negative inotropism, assessed as inhibition of post-rest contractions, in rat right ventricle strips exposed to DOX or to analogues forming fewer amounts of their alcohol metabolites (epirubicin, EPI, and the novel disaccharide anthracycline MEN 10755).
Thirty μM EPI exhibited higher uptake than equimolar DOX, but formed comparable amounts of alcohol metabolite due to its resistance to carbonyl reduction. MEN 10755 exhibited also an impaired uptake, and consequently formed the lowest levels of alcohol metabolite. Accordingly, DOX and EPI inhibited post-rest contractions by ∼40 – 50%, whereas MEN 10755 inhibited by ∼6%.
One hundred μM EPI exhibited the same uptake as equimolar DOX, but formed ∼50% less alcohol metabolite. One hundred μM MEN 10755 still exhibited the lowest uptake, forming ∼60% less alcohol metabolite than EPI. Under these conditions DOX inhibited post-rest contractions by 88%. EPI and MEN 10755 were ∼18% (P<0.05) or ∼80% (P<0.001) less inhibitory than DOX, respectively.
The negative inotropism of 30 – 100 μM DOX, EPI, or MEN 10755 correlated with cellular levels of both alcohol metabolites (r=0.88, P<0.0001) and carbonyl anthracyclines (r=0.79, P<0.0001). Nonetheless, multiple comparisons showed that alcohol metabolites were ∼20 – 40 times more effective than carbonyl anthracyclines in inhibiting contractility. The negative inotropism of MEN 10755 was therefore increased by chemical procedures, like side chain valeryl esterification, that facilitated its uptake and conversion to alcohol metabolite but not its retention in a carbonyl form.
These results demonstrate that secondary alcohol metabolites are important mediators of cardiotoxicity. A combination of reduced uptake and limited conversion to alcohol metabolite formation might therefore render MEN 10755 more cardiac tolerable than DOX and EPI.
Keywords: Doxorubicin, epirubicin, MEN 10755, secondary alcohol metabolites, cardiotoxicity
Introduction
The anthracycline doxorubicin (DOX) is active against several human tumours but also causes severe cardiac toxicity, resulting in heart failure (Weiss, 1992). DOX is composed of aglyconic and sugar moieties. The aglycone, called doxorubicinone, is composed of a quinone-containing tetracyclic ring, and of a short side chain with a carbonyl group at C-13 and a primary alcohol at C-14; the sugar, called daunosamine, is attached by a glycosidic bond to C-7 of the tetracyclic ring, and consists of a 3-amino-2,3,6-trideoxy-L-fucosyl moiety (Figure 1). Two-electron reduction of the side chain carbonyl group (−CO-CH2OH→ −CHOH-CH2OH), mediated by cytoplasmic aldo/keto or carbonyl reductases, converts DOX to a secondary alcohol metabolite (doxorubicinol, DOXol) that was shown to inactivate cytoplasmic aconitase, an important post-transcriptional regulator of iron homeostasis (Minotti et al., 1998). DOXol was also shown to be 10 – 50 times more potent than DOX at inhibiting calcium and proton pumps in isolated sarcoplasmic reticulum or mitochondria (Olson & Mushlin, 1990). Other in vitro studies have questioned the greater toxicity of DOXol vs DOX, and have shown that DOX may be superior to DOXol in inducing inappropriate activation of the calcium release channel (ryanodine receptor) of sarcoplasmic reticulum (Zucchi et al., 2000), or in producing oxygen free radicals through one-electron redox cycling of the quinone moiety (Gervasi et al., 1986). Attempts to recapitulate the different actions of DOX and DOXol in a unifying picture are made difficult by conceptual and technical problems. The myocardial levels of unmetabolized DOX often exceed those of DOXol several-fold (Mushlin et al., 1993), making it difficult to establish whether DOXol reaches sufficient levels for competing or cooperating with DOX in impairing contractility. Inhibitors of aldo/keto or carbonyl reductases, like phenobarbital or rutin-type flavonoids, have been reported to mitigate cardiotoxicity induced by DOX in rats or mice (Behnia & Boroujerdi, 1999; van Acker et al., 1995); however, the decisive role of inhibiting DOXol formation may have been confounded by several other effects of barbiturates and flavonoids, including interferences with free radicals formation and/or reactivity (Almaas et al., 2000; Pietta, 2000). Other problems are posed by the chemical characteristics of DOXol, which is more polar than DOX and consequently exhibits a reduced diffusion from extracellular fluids inside cardiomyocytes (Danesi et al., 1988). This factor limits the interpretation of studies in which isolated cardiac preparations were exposed to DOXol to see how it compared to DOX in impairing contractility (Mushlin et al., 1993). Increased polarity also reduces the cardiac elimination of DOXol as compared to DOX, causing a greater accumulation of the anthracycline molecule inside cardiomyocytes. This factor became evident in studies of transgenic mice bearing cardiac-restricted overexpression of carbonyl reductases. While producing DOXol and developing cardiotoxicity more rapidly than wild type controls, these animals also presented higher cardiac levels of the anthracycline (Forrest et al., 2000). This raises the possibility that cardiotoxicity was enhanced not only by a facilitated formation and action of DOXol, but also by a reduced elimination of the anthracycline molecule from the heart.
Figure 1.
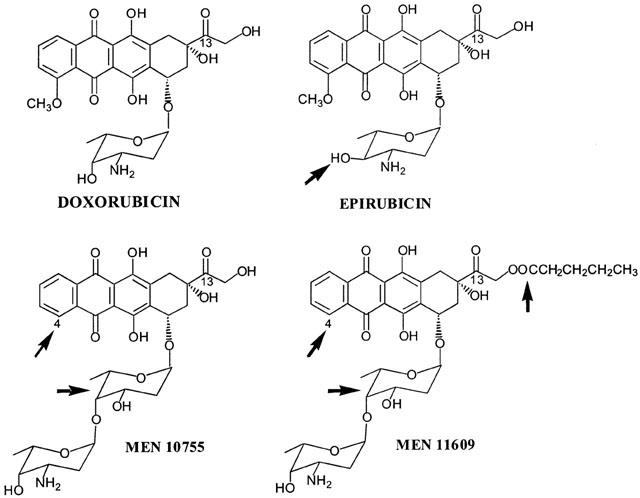
Structures of the anthracyclines used in this study. The arrows indicate epimerization at C-4 in daunosamine (EPI); lack of a methoxy group at C-4 in the aglycone, and intercalation of 2,6-dideoxy-L-fucose between the aglycone and daunosamine (MEN 10755 and MEN 11609); valeryl esterification at C-14 (MEN 11609).
Recent studies in cytosolic fractions from human myocardial samples have allowed us to demonstrate that certain analogues of DOX may be inherently resistant to the action of NADPH-dependent carbonyl reductases (Minotti et al., 2000). In the case of epirubicin (EPI), an approved anthracycline, the formation of its alcohol metabolite EPIol was impaired by axial-to-equatorial epimerization of the hydroxyl group at C-4 in daunosamine. In the case of MEN 10755, an investigational anthracycline, alcohol metabolite formation was diminished by the lack of a methoxy group at C-4 in the aglycone and by the presence of a disaccharide moiety, obtained through intercalation of 2,6-dideoxy-L-fucose between the aglycone and daunosamine (Figure 1). Based upon these findings we reasoned that comparing DOX to EPI or MEN 10755 might be an alternative approach to probing the role of secondary alcohol metabolites as mediators of cardiotoxicity. We therefore designed experiments in which rat isolated heart was exposed to DOX and EPI or MEN 10755, or its valeryl ester MEN 11609 (Figure 1), and the decline in contractility (measured as inhibition of post-rest contraction) was correlated to the cellular levels attained by each anthracycline in its carbonyl or secondary alcohol forms.
Methods
Anthracycline-dependent inhibition of myocardial contractility
Right ventricle strips (∼10 mm long and 2 mm wide) from male Sprague-Dawley rats (∼400 – 450 g) were mounted in a 5 ml organ bath containing 95% O2-5% CO2 Krebs solution, 37°C. These preparations were subjected to continuous electrical-field stimulation with square wave pulses of threshold voltage at 2.5 Hz, using a pair of platinum electrodes. Resting tension was adjusted to 0.5 g and the isometric force of contraction was continuously recorded by using an isometric force transducer and a Basile 7050 polygraph recorder (Varese, Italy). After 60 min equilibration, electrical stimulation was stopped and resumed 30 s later. The amplitude of the first twitch recorded under such conditions is hereafter defined as post-rest contraction (Hagane et al., 1988). Reproducible post-rest contractions were obtained at 5 min intervals; thereafter, anthracyclines were included and their effects on the amplitude of post-rest contractions were determined up to 120 min, a time which was long enough to unravel the effects of both carbonyl anthracyclines and alcohol metabolites (Mushlin et al., 1993), while not impairing contractility in control samples (evidenced by a 98% amplitude of post rest-contractions at the end of the experiments). DOX, EPI and MEN 10755 were used at 30 and 100 μM; MEN 11609 was used at 30 μM due to solubility limit. The effects of anthracyclines were quantified based upon reductions in the amplitude of post-rest contractions, and were expressed as per cent of inhibition relative to controls. At the end of experiments, the ventricle strips were frozen and stored at −80°C until assays for drug content and composition.
Assays for anthracyclines
Ventricle strips were washed in 2 ml of ice-cold 0.3 M NaCl, pH 7.0, homogenized and extracted with a 4 fold excess of (1 : 1) CHCl3/CH3OH. The organic phases were recovered, pooled, and dried under vacuum. Dry residues were dissolved in 200 μl of methanol and cleared of gross particulates by low speed centrifugation. The supernatants were vacuum-dried and residues were suspended in 20 – 50 μl of methanol for analysis by two dimensional TLC on 0.25 mm (20×20 cm) Silica Gel F254 plates (Merck, Darmstadt, Germany) (Minotti et al., 1995; 2000). Mobile phases and Rf values were: CHCl3/CH3OH/CH3COOH/H2O (80 : 20 : 14 : 6) in either dimension for separation of DOX (0.61) and DOXol (0.43); CHCl3/CH3OH/CH3COOH (80 : 20 : 12) in either dimension for separation of EPI (0.50) and EPIol (0.27); CHCl3/CH3OH/CH3COOH/H2O (80 : 20 : 20 : 6) in either dimension for separation of MEN 10755 (0.49), MEN 10755ol (0.35), MEN 11609 (0.72) and MEN 11609ol (0.59). After identification by co-chromatography against authentic standards, parent anthracyclines and secondary alcohol metabolites were eluted and quantified by fluorescence spectroscopy (Minotti et al., 1995). Values were expressed as nmol g−1 (wet weight). Other metabolites, like 7-hydroxy- or deoxy- aglycones, usually were absent or detected at negligible levels; therefore, unless otherwise indicated, total anthracycline uptake was determined based on the sum ((parent anthracycline)+(secondary alcohol metabolite)).
Anthracycline metabolism in human heart
Where indicated, NADPH-dependent conversion of anthracyclines to secondary alcohol metabolites was assayed in cytosolic fractions of human myocardial samples, precisely as reported (Licata et al., 2000; Minotti et al., 2000). Values were expressed as nmol alcohol metabolite mg prot.−1 4 h−1. All myocardial samples derived from the lateral aspect of excluded right atrium of patients undergoing aorto-coronary bypass grafting (Minotti et al., 1995). The specimens were routinely disposed of by the surgeons during cannulation procedures; therefore, patients were not subjected to any unjustified or ethically unacceptable loss of tissue.
Statistical analyses
All values were means±s.e. of 3 – 5 experiments. Differences between two sets of data were analysed by one-tailed unpaired Student's t-test; differences between >2 sets of data were analysed by one-way ANOVA followed by Bonferroni's test for multiple comparisons. Correlations between inhibition of post-rest contractions and anthracycline content and composition were determined by one-tailed nonparametric Spearman analysis. Differences and correlations were considered to be significant when P<0.05.
Drugs
DOX and EPI were obtained through the courtesy of Dr Antonino Suarato (Chemistry Department, Pharmacia-Upjohn, Milan, Italy). MEN 10755 and MEN 11609 were synthesized at the Chemistry Department of Menarini Ricerche S.p.A. (Pomezia, Rome, Italy). Alcohol metabolites were prepared by us after NaBH4 reduction of the side chain carbonyl group (Minotti et al., 1995). All other chemicals were from Sigma (St. Louis, MO, U.S.A.).
Results
Anthracycline uptake, metabolism, and negative inotropism
DOX, EPI and MEN 10755 were evaluated at 30 and 100 μM, concentrations that previous studies have shown to be appropriate for characterizing the pharmacological behaviour of anthracyclines in isolated cardiac preparations (Hagane et al., 1988; Temma et al., 1993). Table 1 shows results obtained at 30 μM. At this concentration DOX exhibited noticeable levels of uptake, retention in its carbonyl form, and conversion to DOXol. EPI exhibited significantly higher levels of uptake and retention than DOX, but the formation of EPIol was similar to that of DOXol. This suggested that the higher uptake and retention of EPI were balanced by the intrinsic resistance of this anthracycline to the action of carbonyl- or aldo/keto- reductases. While exhibiting different uptake and retention but comparable conversion to alcohol metabolites, DOX and EPI proved similar in inhibiting post-rest contractions, an established index of contractility that reflects the amount of calcium available in the sarcoplasmic reticulum (Hagane et al., 1988). This suggested that alcohol metabolites were important determinants of negative inotropism induced by anthracyclines. Interestingly, MEN 10755 was found to produce even less alcohol metabolite than EPI, in spite of the fact that MEN 10755 and EPI exhibited the same resistance to carbonyl reduction when assessed in cell-free systems (Minotti et al., 2000). The reduced formation of MEN 10755ol vs EPIol was attributable to an impaired uptake of MEN 10755 in the ventricle strips, probably reflecting sterical interferences caused by the presence of a disaccharide moiety in the anthracycline molecule. Because of multiple reductions in uptake, retention, and alcohol metabolite formation, MEN 10755 inhibited post-rest contractions by as little as ∼6% as compared to 43 or 50% in the case of DOX or EPI, respectively. Table 2 shows results obtained at 100 μM. This concentration allowed DOX to reach much higher levels of uptake, retention in its carbonyl form, and conversion to DOXol. EPI was able to reach the same levels of uptake and retention, but failed to produce significantly more EPIol than it did at 30 μM, in keeping with its resistance to the action of carbonyl- aldo/keto- reductases. Therefore, EPIol was significantly lower than DOXol when EPI and DOX were assessed at 100 μM. EPI was also slightly but significantly less effective than DOX in inhibiting post-rest contractions, confirming that alcohol metabolites were important determinants of negative inotropism. Under comparable conditions MEN 10755 still exhibited significantly lower levels of uptake, retention, and conversion to alcohol metabolite, and consequently inhibited post-rest contractions much less effectively than DOX or EPI. Collectively, the three anthracyclines impaired myocardial contractility by an order of efficacy that was DOX=EPI > MEN 10755 at 30 μM, or DOX > EPI > MEN 10755 at 100 μM. This pattern of cardiotoxicity seemed to reflect the levels of alcohol metabolites produced by each anthracycline at the two concentrations. However, inhibition of post-rest contractions by 30 – 100 μM DOX, EPI, or MEN 10755, was shown to correlate not only with anthracycline uptake and conversion to alcohol metabolites, but also with cellular retention of anthracyclines in their carbonyl forms (Figure 2). This demonstrated that both carbonyl anthracyclines and secondary alcohol metabolites were involved in impairing contractility.
Table 1.
Anthracycline content, composition, and negative inotropism in rat right ventricle strips: studies at 30 μM

Table 2.
Anthracycline content, composition, and negative inotropism in rat right ventricle strips: studies at 100 μM

Figure 2.
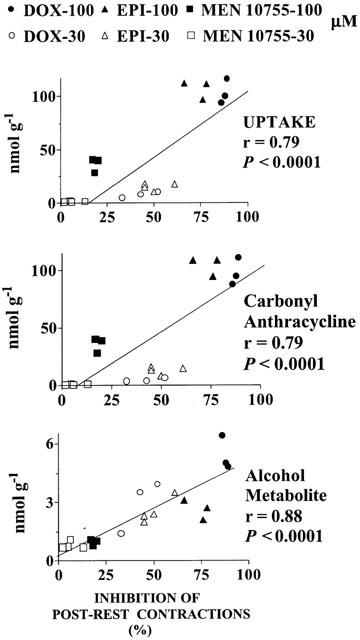
Inhibition of post-rest contractions by DOX, EPI or MEN 10755: Correlations with uptake, carbonyl anthracycline retention, and formation of alcohol metabolites. Values were taken from individual experiments at 30 or 100 μM anthracyclines.
Role of secondary alcohol metabolites
We designed experiments to elucidate the role of alcohol metabolites. For this purpose we used MEN 11609, an analogue obtained by esterifying the side chain C-14 primary alcohol of MEN 10755 with a valeryl residue (cfr. Figure 1). When assessed in human cardiac cytosol, a model which we have developed for screening the structural determinants of anthracycline carbonyl reduction (Licata et al., 2000; Minotti et al., 2000), 30 μM MEN 11609 was similar to equimolar EPI or MEN 10755 in producing less alcohol metabolite than DOX (nmol mg protein−1 4 h−1: DOXol, 0.47±0.06; EPIol, 0.23±0.04; MEN 10755ol, 0.25±0.03; MEN 11609ol, 0.27±0.05; n=3, P<0.05 for EPIol, MEN 10755ol and MEN 11609ol vs DOXol). These findings showed that the presence of a valeryl moiety did not affect the metabolic behaviour of MEN 11609 as compared to authentic MEN 10755. However, the lipophilic character of the valeryl residue was expected to improve diffusion of MEN 11609 in cardiomyocytes, favouring its greater conversion to alcohol metabolite as compared to MEN 10755. Increased lipophilicity was expected to facilitate also the diffusion of unmetabolized MEN 11609 from cardiomyocytes back to the extracellular environment, while leaving the more polar alcohol metabolite inside the cell. MEN 11609 was therefore exploited to increase anthracycline uptake and alcohol metabolite formation while also preventing retention of the carbonyl form of the drug, making it possible to highlight the role of alcohol metabolites in impairing contractility. Incubation of ventricle strips with MEN 11609 resulted in formation of cellular levels of both unmodified drug and MEN 11609ol (3.9±0.3 and 2.1±0.5 nmol g−1 2 h−1, respectively; n=4). Minor amounts of MEN 11609 were recovered also in the form of MEN 10755 and MEN 10755ol (1.2±0.2 and 0.6±0.1 nmol g−1 2 h−1, respectively, n=4), reflecting the activity of esterases which removed the valeryl moiety. Whereas direct measurements of uptake were precluded by continuous equilibration of MEN 11609 across the plasma membrane of cardiomyocytes, carbonyl anthracycline retention could be determined from the sum ((MEN 11609)+(MEN 10755)), and alcohol metabolite formation could be determined from the sum ((MEN 11609ol)+(MEN 10755ol)). These values were compared to those determined for authentic MEN 10755. As shown in Table 3, 30 μM MEN 11609 produced cellular levels of carbonyl anthracycline which were ∼5 times higher than those produced by 30 μM MEN, but ∼7 times lower than those produced by 100 μM MEN 10755. Such differences were statistically significant in the case of 30 μM MEN 11609 vs 100 μM MEN 10755, but not in the case of 30 μM MEN 11609 vs 30 μM MEN 10055. At the same time 30 μM MEN 11609 produced cellular levels of its alcohol metabolite that exceeded ∼3 times, in a statistically significant manner, those produced by either 30 or 100 μM MEN 10755. Such greater levels of conversion to alcohol metabolite, but not of retention in its carbonyl form, enabled 30 μM MEN 11609 to inhibit post-rest contractions ∼5 or ∼2 times more effectively than 30 or 100 μM MEN 10755, respectively, although the increase in negative inotropism of 30 μM MEN 11609 vs 100 μM MEN 10755 did not approach a level of statistical significance when analysed within multiple comparisons (cfr. Table 3). The implications of these findings were several fold. Comparisons between 30 μM MEN 11609 and equimolar MEN 10755 showed that the cardiotoxicity of an anthracycline could be enhanced by facilitating its conversion to alcohol metabolite while not promoting overt increase in retention of its carbonyl form. This demonstrated that alcohol metabolites were independent mediators of anthracycline-induced cardiotoxicity. On the other hand, comparisons between 30 μM MEN 11609 and 100 MEN 10755 showed that the cardiotoxicity of an anthracycline did not decrease, but actually tended to increase 2 fold when a loss in retention of ∼31 nmol of carbonyl anthracycline g−1 was balanced by a net increase of as little as ∼1.7 nmol of alcohol metabolite g−1 (cfr. Table 3). This demonstrated that alcohol metabolites were ∼20 – 40 times more effective than their parent compounds in impairing contractility. Accordingly, the negative inotropism of 30 μM MEN 11609 vs 30 or 100 μM MEN 10755 was shown to correlate in a highly significant manner with the levels of alcohol metabolites but not of carbonyl anthracyclines (Figure 3).
Table 3.
Comparisons between MEN 10755 and MEN 11609

Figure 3.
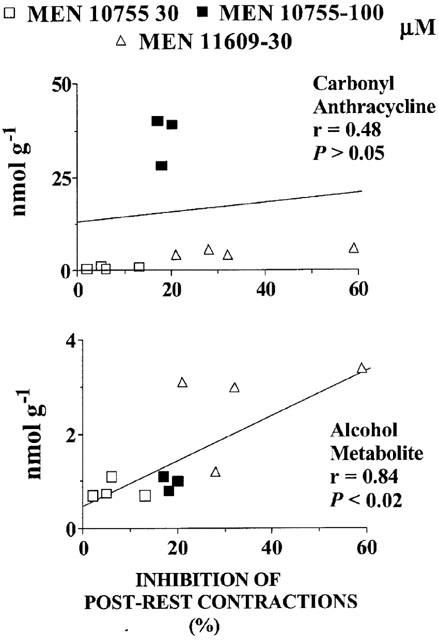
Inhibition of post-rest contractions by MEN 11609 or MEN 10755: Correlations with carbonyl anthracycline retention, and formation of alcohol metabolites. Values were taken from individual experiments at 30 or 100 μM anthracyclines. See also text and Table 3.
Role of carbonyl anthracyclines
To characterize the role of carbonyl anthracyclines we re-analysed experiments in which the drugs produced comparable cellular levels of their alcohol metabolites but exhibited different degrees of inhibition of post-rest contractions. One opportunity was offered by comparisons between 30 and 100 μM MEN 10755. As shown in Figure 4, these concentrations of MEN 10755 resulted in very similar levels of formation of MEN 10755ol inside cardiomyocytes (0.8±0.1 vs 1±0.1 nmol g−1 2 h−1), yet 100 μM MEN 10755 was significantly more effective in inhibiting post-rest contractions, at least when assessed by unpaired Student's t-test for limited comparisons between two groups of data (P<0.001). Because 100 μM MEN 10755 also exhibited a much higher level of retention in its carbonyl form, these observations suggested that carbonyl anthracyclines were able to induce cardiotoxicity in addition to that caused by alcohol metabolites. Nonetheless, it was worth noting that the enhanced negative inotropism of 100 vs 30 μM MEN 10755 required the accumulation of as many as ∼36 nmol of carbonyl anthracycline over ∼1 nmol of alcohol metabolite, confirming that carbonyl anthracyclines were much less toxic than alcohol metabolites. Additional information was obtained by multiple comparisons within a broader panel of anthracyclines (30 μM MEN 11609, 30 μM DOX, and 30 or 100 μM EPI) (Figure 5). These anthracyclines produced fully comparable levels of alcohol metabolites (range: 2.5 – 2.9 nmol g−1 2 h−1). In the case of 30 μM MEN 11609 and DOX, comparable levels of alcohol metabolites coincided with comparable levels of retention of carbonyl anthracyclines (5.0±0.4 and 4.8±0.9 nmol g−1 2 h−1), and of inhibition of post-rest contractions (35±8 vs 43±6%, P>0.05). Thirty μM EPI exhibited levels of carbonyl anthracycline retention that were ∼2.5 times higher than those of 30 μM MEN 10755 or DOX (12.8±1.6 nmol g−1 2 h−1), but these levels were insufficient to produce a significantly increased inhibition of post-rest contractions (50±4%, P>0.05 vs 30 μM MEN 11609 or DOX). Only 100 μM EPI proved to induce a significant exacerbation of negative inotropism (P<0.01 vs the other anthracyclines), but its enhanced toxicity seemed to require cardiac retention of as many as 104±5 nmol of carbonyl anthracycline g−1 2 h−1. These observations confirmed that an accumulation of carbonyl anthracyclines over secondary alcohol metabolites resulted in an increased inhibition of post-rest contractions. However, the observation that such greater negative inotropism required the accumulation of ∼100 nmol of carbonyl anthracycline over ∼2.5 nmol of secondary alcohol metabolite also confirmed that the former was approximately ∼40 times less toxic than the latter.
Figure 4.
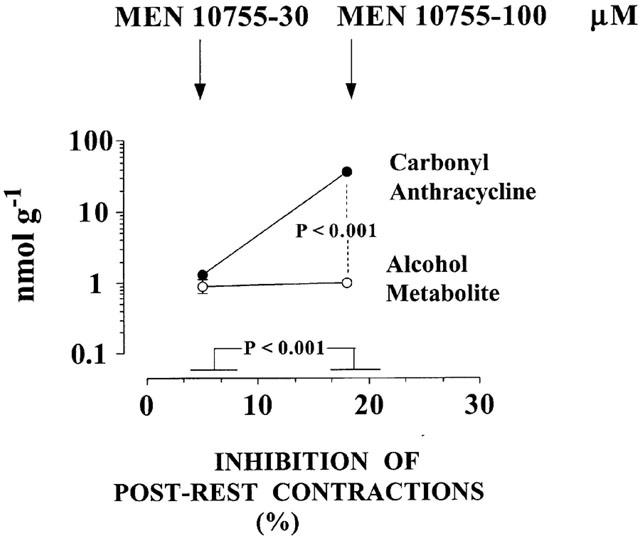
Effects of increasing carbonyl anthracyclines over secondary alcohol metabolites on inhibition of post-rest contractions: Comparisons between 30 and 100 μM MEN 10755. Based on data in Tables 1 and 2, and analysed by unpaired Student's t-test. Values without vertical bars have s.e. within symbols. See also text for explanations.
Figure 5.
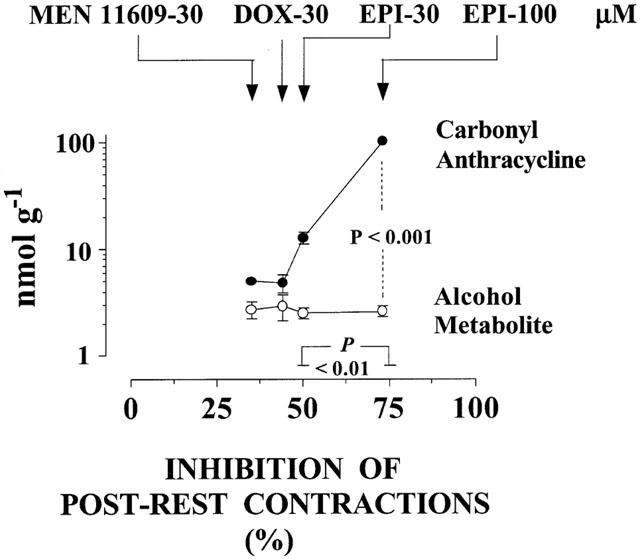
Effects of increasing carbonyl anthracyclines over secondary alcohol metabolites on inhibition of post-rest contractions: Comparisons between 30 μM MEN 11609 or DOX, and 30 – 100 μM EPI. Based on data in Tables 1, 2, 3, and analysed by one-way ANOVA followed by Bonferroni's test for multiple comparisons. Values without vertical bars have s.e. within symbols. See also text for explanations.
Discussion
This study lends support to the concept that secondary alcohol metabolites may be important determinants of cardiotoxicity induced by anthracyclines. This is indicated by several lines of evidence. First, we have shown that 30 μM EPI exhibited higher levels of uptake and retention than equimolar DOX in ventricle strips, yet it did not induce a higher inhibition of post rest-contractions. This was due to the fact that EPI was inherently resistant to the action of carbonyl- or aldo/keto-reductases, and consequently produced as much metabolite as DOX in spite of its improved availability to cellular reductases (cfr. Table 1). Second, we have shown that 100 μM DOX and EPI exhibited comparable levels of uptake and retention, but DOX was significantly more effective than EPI at inhibiting post-rest contractions. This was due to the fact that raising anthracyclines from 30 to 100 μM enhanced the conversion of DOX to DOXol but had no effect in increasing conversion of EPI to EPIol (cfr. Table 2). Third, we have shown that MEN 10755, which is similar to EPI in its intrinsic resistance to alcohol metabolite formation, was less toxic than EPI at both 30 and 100 μM. This was due to the fact that MEN 10755 exhibited also an impaired uptake in the ventricle strips, a factor which diminished formation of MEN 10755ol below the levels attained by EPIol (cfr. Tables 1 and 2). The cardiotoxicity of MEN 10755 was therefore enhanced by chemical modifications, like valeryl esterification in MEN 11609, which increased its uptake and conversion to alcohol metabolite. Our results also demonstrate that the cardiotoxicity of secondary alcohol metabolites cannot be attributed to increased polarity and consequent retention of a larger pool of the anthracycline molecule inside cardiomyocytes. In fact, the greater negative inotropism of 30 μM MEN 11609 vs 30 or 100 μM MEN 10755 was observed under conditions in which a facilitated formation of the alcohol metabolite was accompanied by either marginal increase or significant decrease in retention of the carbonyl form of the anthracycline, respectively, making negative inotropism eventually correlate with the levels of alcohol metabolites but not of their parent compounds (cfr. Table 3 and Figure 3).
Anthracycline-induced cardiotoxicity is a multifactorial process in which also the carbonyl forms of the drugs can contribute to impair contractility. This information has been obtained by comparing groups of anthracyclines which produced comparable cellular levels of their alcohol metabolites but exhibited different negative inotropism and retention in their carbonyl forms (cfr. Figures 4 and 5). However, a quantitative analysis of such comparisons suggests that carbonyl anthracyclines may be ∼40 times less active than their alcohol metabolites in inhibiting post-rest contractions. The same approximation was reached in the experiments in which MEN 11609 was exploited for the opposite purpose of increasing the levels of alcohol metabolite formation while not facilitating retention of the carbonyl anthracycline. Understanding that anthracycline-induced cardiotoxicity is a multifactorial process, involving carbonyl and alcohol species, has several important implications. On the one hand, this picture helps to explain how the negative inotropism of 30 – 100 μM DOX, EPI, and MEN 10755 correlates with the cardiac levels of both carbonyl anthracyclines and secondary alcohol metabolites. On the other hand, the much greater toxicity of alcohol metabolites vs carbonyl anthracyclines helps to explain how the metabolites always contribute to impair contractility, even in the presence of a ∼20 – 40 fold excess of their parent compounds (e.g., in ventricle strips exposed to 100 μM anthracyclines) (cfr. Table 2 and Figure 2).
An intriguing feature of secondary alcohol metabolites is that they do not always contribute to the antitumour activity of anthracyclines (Kuffel et al., 1992); in some cases, an excessive conversion to secondary alcohol metabolites actually diminishes the antitumour efficacy of anthracyclines (Gonzalez et al., 1995; Ax et al., 2000). Here we have demonstrated that secondary alcohol metabolites may be several fold more effective than their parent anthracyclines in inducing cardiotoxicity. The differential actions of secondary alcohol metabolites in cancer cells or cardiomyocytes are therefore exploitable for designing anthracyclines which form fewer amounts of such metabolites and consequently exhibit equal or improved antitumour activity, while also inducing less severe cardiac toxicity. Clinical studies have confirmed that EPI, characterized by an intrinsic resistance to conversion to its alcohol metabolite, retains good antitumour activity but causes heart failure only after administration of higher cumulative doses than DOX (Weiss, 1992). Similarly, preclinical studies have shown that MEN 10755, whose conversion to MEN 10755ol is limited also by a reduced uptake in cardiac tissue, induces less severe cardiotoxicity when given chronically to rats (Cirillo et al., 2000), but exhibits greater activity than DOX in tumour cell lines or human tumour xenografts (Arcamone et al., 1997; Pratesi et al., 1998). Recent studies in athymic mice xenografted with human ovarian cancer and treated with a single i.v. dose of anthracyclines have shown that MEN 10755 exhibited a reduced uptake not only in normal tissues but also in the tumour, yet it retained greater therapeutic efficacy than DOX (Gonzalez-Paz et al., 2001). This suggests that MEN 10755 is intrinsically superior to DOX in inducing topoisomerase II inhibition and apoptosis in cancer cells (Arcamone et al., 1997; Pratesi et al., 1998), consistent with its impaired conversion to an alcohol metabolite exhibiting reduced activities in these settings.
In conclusion, we have shown that secondary alcohol metabolites are important mediators of cardiotoxicity induced by anthracyclines, and that a unique combination of reduced uptake and alcohol metabolite formation might improve the cardiac safety of MEN 10755 in comparison to DOX or EPI. According to in vitro studies, the cardiotoxicity of MEN 10755 would be further reduced by the fact that disaccharide secondary alcohol metabolites are less reactive than DOXol or EPIol toward cytoplasmic aconitase (Minotti et al., 2000). This peculiar aspect does not seem to have surfaced in the present study; in fact, comparisons between 30 μM DOX, EPI, and MEN 11609, which produced comparable cellular levels of alcohol metabolites within a narrow range of retention of their carbonyl forms, clearly showed that the three anthracyclines were similar in inhibiting post-rest contractions (cfr. Figure 5). The reduced reactivity of disaccharide metabolites might nonetheless surface, and prove useful to further prevent cardiotoxicity, if anthracyclines were given chronically and cytoplasmic aconitase were needed to counteract alterations of iron homeostasis induced by the presence of cancer and inflammatory reactions (Torti & Torti, 1994; Minotti et al., 1996). Whereas the mechanisms of alcohol metabolite-induced toxicity remain open to debate (Minotti et al., 1999), the results described in this study, and previous evidence for a higher antitumour activity of MEN 10755 vs DOX, anticipate that MEN 10755 might exhibit a better therapeutic index than other approved anthracyclines.
Acknowledgments
This work was supported by AIRC; MURST COFIN '99; and MURST “Center of Excellence in Aging at the University of Chieti” (to G. Minotti).
Abbreviations
- Anthracycline-ol
anthracycline secondary alcohol metabolite
- DOX
doxorubicin (7-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)]doxorubicinone)
- DOXol
doxorubicinol (C-13 hydroxydoxorubicin)
- EPI
epirubicin (7-[(3-amino-2,3,6-trideoxy-α-L-arabino-hexopyranosyl)]doxorubicinone)
- MEN 10755
(7-[(2,6-dideoxy-4-O-[3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)]-α-L-lyxohexopyranosyl]-4-demethoxydoxorubicinone)
- MEN 11609
(7-[(2,6-dideoxy-4-O-[3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)]-4-demethoxy-14-O-valeryl-doxorubicinone)
References
- ALMAAS R., SAUGSTAD O.D., PLEASURE D., ROOTWELT T. Effect of barbiturates on hydroxyl radicals, lipid peroxidation, and hypoxic cell death in human NT2-N neurons. Anesthesiology. 2000;92:764–774. doi: 10.1097/00000542-200003000-00020. [DOI] [PubMed] [Google Scholar]
- ARCAMONE F., ANIMATI F., BERETTONI M., BIGIONI M., CAPRANICO G., CASAZZA A.M., CASERINI C., CIPOLLONE A., DE CESARE M., FRANCIOTTI M., LOMBARDI P., MADAMI A., MANZINI S., MONTEAGUDO E., POLIZZI D., PRATESI G., RIGHETTI S.C., SALVATORE C., SUPINO R., ZUNINO F. Doxorubicin disaccharide analogue: apoptosis-related improvement of efficacy in vivo. J. Natl. Cancer Inst. 1997;89:1217–1223. doi: 10.1093/jnci/89.16.1217. [DOI] [PubMed] [Google Scholar]
- AX W., SOLDAN M., KOCH L., MASER E. Development of daunorubicin resistance in tumour cells by induction of carbonyl reduction. Biochem. Pharmacol. 2000;59:293–300. doi: 10.1016/s0006-2952(99)00322-6. [DOI] [PubMed] [Google Scholar]
- BEHNIA K., BOROUJERDI M. Inhibition of aldo-keto reductases by phenobarbital alters metabolism, pharmacokinetics and toxicity of doxorubicin in rats. J. Pharm. Pharmacol. 1999;51:1275–1282. doi: 10.1211/0022357991777010. [DOI] [PubMed] [Google Scholar]
- CIRILLO R., SACCO G., VENTURELLA S., BRIGHTWELL J., GIACHETTI A., MANZINI S. Comparison of doxorubicin- and MEN 10755-induced long term progressive cardiotoxicity in the rat. J. Cardiov. Pharmacol. 2000;35:100–108. doi: 10.1097/00005344-200001000-00013. [DOI] [PubMed] [Google Scholar]
- DANESI R., PAPARELLI A., BERNARDINI N., DEL TACCA M. Cytofluorescence localization and disposition of doxorubicin and doxorubicinol in rat cardiac tissue. Eur. J. Cancer Clin. Oncol. 1988;24:1123–1131. doi: 10.1016/0277-5379(88)90118-6. [DOI] [PubMed] [Google Scholar]
- FORREST G.L., GONZALEZ B., TSENG W., LI X., MANN J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res. 2000;60:5158–5164. [PubMed] [Google Scholar]
- GERVASI P.G., AGRILLO M.R., CITTI L., DANESI R., DEL TACCA M. Superoxide anion production by adriamycinol from cardiac sarcosomes and by mitochondrial NADH dehydrogenase. Anticancer Res. 1986;6:1231–1235. [PubMed] [Google Scholar]
- GONZALEZ B., AKMAN S., DOROSHOW J., RIVERA H., KAPLAN W.D., FORREST G.L. Protection against daunorubicin cytotoxicity by expression of a cloned human carbonyl reductase cDNA in K562 leukemia cells. Cancer Res. 1995;55:4646–4650. [PubMed] [Google Scholar]
- GONZALEZ-PAZ O., POLIZZI D., DE CESARE M., ZUNINO F., BIGIONI M., MAGGI C.A., MANZINI S., PRATESI G. Tissue distribution, antitumour activity and in vivo apoptosis induction by MEN 10755 in nude mice. Eur. J. Cancer. 2001;37:431–437. doi: 10.1016/s0959-8049(00)00414-7. [DOI] [PubMed] [Google Scholar]
- HAGANE K., AKERA T., BERLIN J.R. Doxorubicin : Mechanism of cardiodepressant actions in guinea pigs. J. Pharmacol. Exptl. Ther. 1988;246:655–661. [PubMed] [Google Scholar]
- KUFFEL M.J., REID J.M., AMES M.M. Anthracyclines and their C-13 alcohol metabolites: growth inhibition and DNA damage following incubation with tumor cell lines. Cancer Chemother. Pharmacol. 1992;30:51–57. doi: 10.1007/BF00686485. [DOI] [PubMed] [Google Scholar]
- LICATA S., SAPONIERO A., MORDENTE A., MINOTTI G. Doxorubicin metabolism and toxicity in human myocardium: role of cytoplasmic deglycosidation and carbonyl reduction. Chem. Res. Toxicol. 2000;13:414–420. doi: 10.1021/tx000013q. [DOI] [PubMed] [Google Scholar]
- MINOTTI G., CAIRO G., MONTI E. Role of iron in anthracycline cardiotoxicity : new tunes for an old song. FASEB J. 1999;13:199–212. [PubMed] [Google Scholar]
- MINOTTI G., CAVALIERE A.F., MORDENTE A., ROSSI M., SCHIAVELLO R., ZAMPARELLI R., POSSATI G.F. Secondary alcohol metabolites mediate iron delocalization in cytosolic fractions of myocardial biopsies exposed to anticancer anthracyclines. J. Clin. Invest. 1995;95:1595–1605. doi: 10.1172/JCI117833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINOTTI G., LICATA S., SAPONIERO A., MENNA P., CALAFIORE A.M., DI GIAMMARCO G., LIBERI G., ANIMATI F., CIPOLLONE A., MANZINI S., MAGGI C.A. Anthracycline metabolism and toxicity in human myocardium: comparisons between doxorubicin, epirubicin, and a novel disaccharide analogue with a reduced level of formation and [4Fe-4S] reactivity of its secondary alcohol metabolite. Chem. Res. Toxicol. 2000;13:1336–1341. doi: 10.1021/tx000143z. [DOI] [PubMed] [Google Scholar]
- MINOTTI G., MANCUSO S., FRUSTACI A., MORDENTE A., SANTINI S.A., CALAFIORE A.M., LIBERI G., GENTILONI N. Paradoxical inhibition of cardiac lipid peroxidation in cancer patients treated with doxorubicin. J. Clin. Invest. 1996;96:650–661. doi: 10.1172/JCI118836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINOTTI G., RECALCATI S., LIBERI G., CALAFIORE A.M., MANCUSO C., PREZIOSI P., CAIRO G. The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB J. 1998;12:541–551. doi: 10.1096/fasebj.12.7.541. [DOI] [PubMed] [Google Scholar]
- MUSHLIN P.S., CUSACK B.J., BOUCEK R.J., JR, ANDREJUK T., LI X., OLSON R.D. Time-related increases in cardiac concentrations of doxorubicinol could interact with doxorubicin to depress myocardial contractile function. Br. J. Pharmacol. 1993;110:975–982. doi: 10.1111/j.1476-5381.1993.tb13909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSON R.D., MUSHLIN P.S. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB J. 1990;4:3076–3086. [PubMed] [Google Scholar]
- PIETTA P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000;63:1035–1042. doi: 10.1021/np9904509. [DOI] [PubMed] [Google Scholar]
- PRATESI G., DE CESARE M., CASERINI C., PEREGO P., DAL BO L., POLIZZI D., SUPINO R., BIGIONI M., MANZINI S., IAFRATE E., SALVATORE C., CASAZZA A., ARCAMONE F., ZUNINO F. Improved efficacy and enlarged spectrum of activity of a novel anthracycline disaccharide analogue of doxorubicin against human tumor xenografts. Clin. Cancer Res. 1998;4:2833–2839. [PubMed] [Google Scholar]
- TEMMA K., AKERA T., CHUGUN A., KONDO H., HAGANE K., HIRANO S. Comparison of cardiac actions of doxorubicin, pirarubicin and aclarubicin in isolated guinea-pig heart. Eur. J. Pharmacol. 1993;234:173–181. doi: 10.1016/0014-2999(93)90951-d. [DOI] [PubMed] [Google Scholar]
- TORTI F.M., TORTI S.V. Cytokines, iron homeostasis, and cancer. Adv. Exp. Med. Biol. 1994;354:161–170. doi: 10.1007/978-1-4899-0939-8_12. [DOI] [PubMed] [Google Scholar]
- VAN ACKER S.A., KRAMER K., GRIMBERGEN J.A., VAN DEN BERG D.J., VAN DER VIJGH W.J., BAST A. Monohydroxyethylrutoside as protector against chronic doxorubicin-induced cardiotoxicity. Br. J. Pharmacol. 1995;115:1260–1264. doi: 10.1111/j.1476-5381.1995.tb15034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS R.B. The anthracyclines: will we ever find a better doxorubicin. Sem. Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- ZUCCHI R., YU G., GHELARDONI S., RONCA F., RONCA-TESTONI S. Effect of MEN 10755, a new disaccharide analogue of doxorubicin, on sarcoplasmic reticulum Ca(2+) handling and contractile function in rat heart. Br. J. Pharmacol. 2000;131:342–348. doi: 10.1038/sj.bjp.0703575. [DOI] [PMC free article] [PubMed] [Google Scholar]