

Abstract
The present study was performed to evaluate and compare the ability of human MDR1-, and rat Mdr1b- and Mdr2-P-glycoproteins to transport hydrophilic monoquaternary drugs. Transport studies were performed with plasma membrane vesicles isolated from MDR1-, Mdr1b-, or Mdr2-overexpressing insect cells.
As model substrates we used the N-methylated derivatives of the diastereomers quinidine and quinine, the monoquaternary compounds N-methylquinidine and N-methylquinine. Vincristine, an established MDR1 substrate, was used as a reference.
We observed ATP-dependent uptake of all drugs studied into MDR1- and Mdr1b-expressing vesicles. Mdr2 was not able to transport these compounds. MDR1- and Mdr1b-mediated transport was saturable, and could be inhibited by various drugs, including PSC-833.
For both MDR1 and Mdr1b the Vmax/Km ratios (or clearance) of N-methylquinidine were greater than those determined for N-methylquinine. This stereoselective difference was also evident from differential inhibitory studies with the two isomers.
Comparison of normalized clearance indicated that human MDR1 was more effective in transporting the tested substrates than rat Mdr1b.
In conclusion, our results demonstrate that MDR1 and Mdr1b, but not Mdr2, are able to transport the monoquaternary model drugs; both MDR1 and Mdr1b display stereospecificity for these cations; and indicate human MDR1 is more efficient in transporting these cations than its rat orthologue Mdr1b.
Keywords: ABC transporter, P-glycoprotein, human MDR1, rat Mdr1b, rat Mdr2, ATP-dependent drug transport, drug secretion, stereoisomers, enzyme kinetics, baculovirus expression system
Introduction
Class 1 P-glycoproteins (Pgps) are ATP-dependent drug transporting membrane proteins, typically associated with multidrug resistance in cancer therapy (Gottesman & Pastan, 1993; Ambudkar et al., 1999). In humans, the drug transporting Pgp is encoded by the MDR1 gene (Chen et al., 1986), whereas in rodents two drug transporting Pgp isoforms have been identified, encoded by the Mdr1a and Mdr1b genes, respectively (Devault & Gros, 1990; Deuchars et al., 1992). The closely related human MDR3 and rodent Mdr2 proteins (class 2 Pgps) do not confer multidrug resistance, but primarily function as phosphatidylcholine flippases (Oude Elferink et al., 1997). Class 1 Pgps confer drug resistance to cells by the active excretion of a variety of structurally unrelated hydrophobic natural product drugs, e.g. vinca alkaloids and anthracyclines, which ultimately results in reduced cytoplasmic drug concentrations and drug-induced toxicity (Gottesman & Pastan, 1993; Ambudkar et al., 1999). Tissue distribution studies revealed that class 1 Pgps are not only expressed in drug resistant cells, but also in the apical domain of cells in normal tissue with excretory functions, such as liver, small intestine, kidney, and at the blood – brain barrier (Thiebaut et al., 1987). Considering the distribution pattern in excretory organs, combined with their capacity to transport drugs, it has been suggested that in mammals class 1 Pgps are involved in the excretion and disposition of amphiphilic substrates. First evidence in support of this hypothesis was obtained by Kamimoto et al. (1989). In studies with rat liver canalicular membrane vesicles they demonstrated that class 1 Pgps may be involved in the hepatobiliary excretion of various drugs. Later studies, in which other substrates or other techniques were employed further supported these observations (Watanabe et al., 1992; Müller et al., 1994a). In the study of Müller et al. (1994a), preliminary evidence was obtained indicating that ATP-dependent transport of the cationic drugs N-(4′,4′-azo-n-pentyl)-21-deoxyajmalinium and N-(n-pentyl)-quinidine into rat canalicular plasma membrane vesicles was mediated by Mdr1 Pgps. However, stereoselective aspects of this transport were not studied.
More recently, experiments with Mdr1 gene knockout mice have shed light on the importance of class 1 Pgps in vivo. Mice with a disruption of the Mdr1 genes grow and develop normally, are fertile, and have no obvious abnormality under laboratory conditions. However, these mice are very sensitive to mdr-related toxic chemicals, including chemotherapeutic drugs, due to profoundly reduced clearance of these agents in liver, intestine and brain (Schinkel, 1998). Interestingly, absence of class 1 Pgps results in a significant reduction in excretion rates not only of typical Pgp substrates, like natural product drugs, but also of aliphatic organic cations (Smit et al., 1998a, 1998b), suggesting that class 1 Pgps are also involved in the elimination of relatively small, permanently-charged cationic drugs. Results from an in vitro study, in which transfected polarized cells were used, support these observations (Smit et al., 1998c). However, it should be noted that in the before-mentioned studies, Pgp-mediated drug transport can only be indirectly inferred. Furthermore, potential influences of the knockout or transfection procedures on transport proteins, other than the Pgps, cannot be entirely excluded in these studies.
In order to provide direct information on Pgp-mediated transport of relatively small, permanently-charged cationic drugs, we have performed uptake studies with membrane vesicle preparations isolated from Pgp-overexpressing cells using quaternary derivatives of the enantiomers quinidine and quinine as model substrates. Specifically, we have investigated whether these cationic drugs are substrates for human MDR1-, rat Mdr1b-, or rat Mdr2-Pgp, and compared the observed kinetic parameters using vincristine transport as a reference. Moreover, by evaluating the transport of two diastereomers we were able to address possible stereoselectivity of class 1 Pgp-mediated transport.
Methods
Materials
[G-3H]-vincristine (5.80 Ci mmol−1) was obtained from Amersham (Little Chalfont, U.K.). The permanently charged cationic compounds [3H]-N-methylquinidine (85 Ci mmol−1) and [3H]-N-methylquinine (85 Ci mmol−1) were synthesized by N-methylation of quinidine and quinine, respectively, with [3H]-methyliodide (Amersham) as described (van montfoort et al., 1999). Unlabeled N-methylquinidine and N-methylquinine were also obtained by N-methylation of quinidine and quinine, respectively. The molecular weight of the model compounds was assessed by mass spectroscopy, confirming the methylation of the quinuclidine nitrogen (van montfoort et al., 1999). Thin-layer chromatography (TLC) was used to determine the radiochemical purity of the non-commercially available substrates. Briefly, samples were spotted on silica gel sheets (Merck, Darmstadt, Germany), and developed in the organic layer of methanol : chloroform : sodium bromide (80 : 20 : 5 v/v/w). Radioactive products were identified using a Berthold Tracemaster 40 Automatic TLC-Linear analyser (Berthold Systems Inc., Pittsburgh, PA, U.S.A.). Retention factors (Rf) were 0.63 and 0.68 for [3H]-N-methylquinidine and [3H]-N-methylquinine, respectively, and the radiochemical purity of the two enantiomers exceeded 98.5%. ATP, creatine phosphate, and creatine kinase were purchased from Roche Molecular Biochemicals (Mannheim, Germany). Vincristine, β,γ-methyleneadenosine 5′ -triphosphate (AMP – PCP) were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). All other chemicals were of analytical grade and readily available from commercial sources. The Bac-to-Bac baculovirus expression system was obtained from Life Technologies (Breda, The Netherlands). Sf21 insect cells were purchased from Invitrogen (Leek, The Netherlands) and cultured in Insect Xpress medium (BioWhittaker, Walkersville, MD U.S.A.) supplemented with 5% foetal bovine serum and 25 μg ml−1 penicillin, 25 μg ml−1 streptomycin and 50 μg ml−1 neomycin (Life Technologies). A mouse monoclonal antibody C219 (Signet Laboratories Inc, Dedham, MA, U.S.A.) was used for detection of P-glycoproteins. Cellulose acetate filters (OE66) were from Schleicher & Schuell (Dassel, Germany), and glass microfibre filters (GF/F) were purchased from Whatman (Maidstone, UK). Molecular biology reagents were obtained from Promega Corporation (Leiden, The Netherlands) or Roche Molecular Biochemicals. Plasmids containing the rat Mdr1b cDNA (Silverman et al., 1991) or rat Mdr2 cDNA (Brown et al., 1993) were generously provided by Dr J.A. Silverman, (AvMax Inc., Berkeley, CA, U.S.A.). Dr U.S. Rao (University of North Carolina, Chapel Hill, U.S.A.) kindly provided an aliquot of a high titre baculovirus stock containing the human MDR1 gene (Rao, 1995).
Production of recombinant baculovirus and viral infection
Recombinant baculoviruses were generated using the Bac-to-Bac baculovirus expression system from Life Technologies, according to the manufacturer's instructions. Briefly, rat Mdr1b and Mdr2 coding sequences were ligated in pFASTBAC1. Clones with the correct orientation of the Mdr1b or Mdr2 cDNA in pFASTBAC1 were identified by restriction analysis and confirmed by DNA sequencing. Recombinant bacmids were created and used for transfection of Sf21 insect cells using Cellfectin reagent. After 5 days culture medium was collected and used for a plaque assay. Single plaques were isolated, eluted into medium and used for three-rounds of amplification to obtain high titer virus stocks. The resulting virus stocks had a titer of 5 – 8 107 pfu ml−1, as determined by plaque assays, and were used for the production of recombinant protein in Sf21 insect cells. The baculovirus stock containing the human MDR1 gene was amplified to obtain large volumes of a high titer virus stock. One day before infection, Sf21 insect cells were seeded at a density of 1.5·107 cells 30 ml−1 medium in 225 cm2 flasks. The next day cells were infected at a multiplicity of infection of 2 – 4 with recombinant baculoviruses. For control experiments, Sf21 insect cells were infected with recombinant baculoviruses containing the β-glucuronidase gene (Life Technologies). Plasma membrane fractions were isolated 65 – 67 hours after infection (see below).
Isolation of plasma membrane fractions
Plasma membrane-enriched fractions were isolated from infected Sf21 insect cells by sucrose gradient centrifugation techniques according to Müller et al. (1994b) with some modifications. Briefly, cells (approximately 1.5 – 2·108) were harvested, washed with phosphate buffered saline (PBS); mM: NaCl 137, KCl 2.7, Na2HPO4 10.1, KH2PO4 1.8, pH 7.4) and centrifuged at 700×g for 10 min at 4°C. The resulting pellet was diluted 50 fold in hypotonic buffer (0.5 mM sodium phosphate) supplemented with 0.1 mM EDTA and 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and stirred gently for 90 min on ice. The cell lysate was centrifuged at 100,000×g for 45 min at 4°C. The pellet of the crude membranes was resuspended in isotonic TS buffer (10 mM Tris-HCl, pH 7.4, 250 mM sucrose) supplemented with 0.1 mM EDTA and 0.1 mM PMSF, and homogenized with a Dounce B homogenizer (50 strokes up-and-down). The suspension was layered on top of a 38% (w/w) sucrose solution and centrifuged at 280,000×g for 2 h at 4°C in a swing out rotor. The interface layer was collected, diluted to 25 ml in TS buffer and centrifuged at 100,000×g for 45 min at 4°C. The resulting pellet was resuspended in 750 μl of isotonic buffer. Vesicles were formed by passing the suspension 25 times through a 25-gauge needle. The membrane vesicles were aliquotted into 35 μl portions, snap frozen in liquid nitrogen and stored at −80°C until use.
Immunoblot blot analysis and quantification of MDR1 and Mdr1b P-glycoproteins in membranes
Plasma membrane-enriched protein fractions were separated by sodium dodecyl sulphate-polyacrylamide (final concentration 7.5%) gel electrophoresis (SDS – PAGE) and transferred to nitrocellulose (Amersham International, Buckinghamshire, U.K.) using a tank-blotting system according to manufacturer's instructions (BioRad Laboratories, Hercules, CA, U.S.A.). BDH molecular weight standards (42,700 – 200,000 Dalton range, BDH Ltd, Dorset, U.K.) were used as marker proteins. The blots were stained with Ponceau S-solution (0.1% Ponceau S (w v−1) in 5% acetic acid (v v−1), Sigma) to confirm equal transfer of proteins. The blots were incubated with the monoclonal antibody C219 (dilution 1 : 2000) in PBS containing 4% non-fat dried milk powder (Fluka BioChemica, Buchs, Switzerland) and 0.05% polyoxyethylene sorbitan monolaurate (Tween-20, Sigma), washed in PBS / 0.05% Tween-20, subsequently incubated with horseradish peroxidase labelled rabbit anti-mouse IgG (dilution 1 : 2000, DAKO A/S, Glostrup, Denmark) and developed using Pierce SuperSignal Chemiluminescent Substrate Luminol/Enhancer (Pierce, Rockford, IL, U.S.A.). The amount of MDR1 or Mdr1b in membrane fractions was determined by quantitative immunoblotting using the monoclonal antibody C219. This antibody recognizes conserved epitopes close to the ATP binding cassette present in all known members of the Pgp-subfamily (Georges et al., 1990). However, the peptide epitopes in human MDR1 and rat Mdr1b bind to C219 antibody with different affinities [data not shown, Georges et al. (1990)]. Thus, it is not possible to directly determine and compare Pgp expression levels solely by immunoblot analysis of equal amounts of Sf-MDR1 and Sf-Mdr1b membrane preparations loaded on the same gel. Briefly, samples were serially diluted in TS buffer. Standard solutions of purified human MDR1 or rat Mdr1b P-glycoprotein with known protein concentration were prepared by electroelution (see below). Samples and standards, containing 350 ng to 25 ng of P-glycoprotein, were separated by SDS – PAGE, transferred to nitrocellulose and stained with C219. Band intensities of the immunoblots were quantified using ImageMaster 1D Elite software, version 3.0 (Pharmacia, Uppsala, Sweden).
Electroelution of MDR1 and Mdr1b
Plasma membrane-enriched protein fractions were separated by SDS – PAGE. After electrophoresis, the positions of MDR1 and Mdr1b were determined by staining of the gel in 0.3 M CuCl2 (Lee et al., 1987). The horizontal strips containing P-glycoproteins were cut out, destained in 250 mM EDTA/250 mM Tris HCl (pH 9.0), minced, and placed into the glass tubes of a BioRad 422 Electro-Eluter Module (BioRad Laboratories). Elution was carried out at 10 mA per tube at constant current for 5 h in 20 mM Tris/150 mM glycine buffer (pH 8.3) containing 0.01% (w v−1) SDS, with membrane caps with a cut-off of 12 – 15 kDa. After elution, protein concentrations of purified samples were determined by densitometric analysis of Coomassie-stained polyacrylamide gels, using BSA as a standard. Images of gels were taken using a charge-coupled device video camera of the ImageMaster VDS system (Pharmacia) and band intensities were quantified using ImageMaster software (Pharmacia).
Transport studies with membrane vesicles
Transport of [3H]-labelled substrates was measured by a rapid filtration technique using cellulose acetate (0.20 μm pore size, Schleicher & Schuell) or glass microfibre filters (0.70 μm pore size, Whatman), according to Renes et al. (1999) with some slight modifications. Briefly, membrane vesicles (25 – 30 μg protein) were rapidly thawed and then preincubated at 37°C in the presence of 4 mM ATP, 10 mM MgCl2, 10 mM creatine phosphate, 100 μg ml−1 of creatine kinase, 250 mM sucrose, 10 mM Tris HCl (pH 7.4). After 1 min the labelled substrate was added (final volume 40 μl). At the indicated time points 1 ml of ice-cold stop solution (PBS) was added to the membranes. This solution was subsequently filtered through cellulose acetate filters (for N-methylquinidine and N-methylquinine) or glass microfibre filters (for vincristine), that were pre-soaked in PBS. Filters were rinsed with 5 ml PBS/1% BSA, followed by 5 ml PBS/0.05% Tween-20 and 5 ml PBS. After rinsing, the filters were air-dried, dissolved, and radioactivity was measured in a liquid scintillation counter. In control experiments ATP was replaced by 4 mM AMP – PCP. Net ATP-dependent transport was calculated by subtracting values obtained in the presence of AMP – PCP from those obtained in the presence of ATP. The total recovery of radioactivity from the solution varied between 5 – 10%, the signal to noise ratio was 15 – 20 : 1.
Distribution coefficient determinations
The relative hydrophilicity of the cationic compounds was analysed by determining the octanol-aqueous phase distribution coefficients (D7.4) from an n-octanol and isotonic TS buffer (pH 7.4) system. Briefly, radiolabelled compounds were dissolved in equal volumes of n-octanol and TS buffer. The two phases were mixed continuously for 2 h at 20°C. The layers were separated by centrifugation at 2500 r.p.m. for 15 min, and analysed in a liquid scintillation counter.
Analytical procedures
Protein concentrations of membrane preparations were determined with the DC Protein Assay from BioRad Laboratories (Hercules, CA, U.S.A.) using BSA as standard. Kinetic uptake parameters were determined by non-linear curve fitting to an equation describing simple Michaelis – Menten enzyme kinetics, v=Vmax×[S]/(Km+[S]), using SigmaPlot 3.03 (Jandel Scientific Corporation, San Rafael, CA, U.S.A.).
Statistical analysis
All data are presented as mean values±standard deviation, unless otherwise stated. Statistical analysis between the experimental groups in the inhibition study was performed using oneway analysis of variance (ANOVA), followed by the Student – Newman – Keuls test. Levels of significance for statistical analysis was set at P<0.05.
Results
Immunoblot analysis of MDR1, Mdr1b, and Mdr2 in membrane vesicles
Expression of MDR1, Mdr1b, and Mdr2 in isolated membrane preparations from infected Sf21 insect cells was examined by immunoblot analysis using the monoclonal antibody C219. For comparison, membranes from A2780/AD cells, a MDR1-overexpressing multidrug resistant human ovarian cell line (Renes et al., 1999) were included in the immunoblot analysis. Human MDR1 was detected in membrane preparations isolated from insect cells infected with a MDR1-expressing recombinant baculovirus (Sf-MDR1 membranes), and in membranes prepared from A2780/AD cells (Figure 1). The apparent molecular weight of MDR1 expressed in insect cells is smaller than MDR1 detected in A2780/AD cells, which can be attributed to differences in post-translational modifications (Germann et al., 1990; Sarkadi et al., 1992). Furthermore, the expression level of human MDR1 in Sf21 cells is approximately four times higher than in A2780/AD cells. In addition, strong C219-immunoreactive signals were observed in membrane preparations from Sf21 cells infected with either rat Mdr1b- or Mdr2-expressing recombinant baculoviruses (Sf-Mdr1b and Sf-Mdr2 membranes, respectively). No C219-immunoreactive signals were observed in non-infected Sf21 insect cells. The amount of MDR1 or Mdr1b present in membrane preparations was determined by quantitative immunoblotting using purified MDR1 or Mdr1b as standards.
Figure 1.
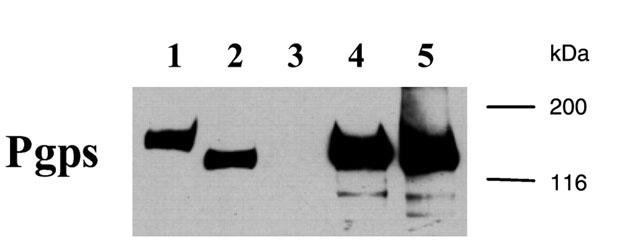
Immunoblot analysis of MDR1, Mdr1b, and Mdr2 in membrane vesicle preparations. Plasma membrane-enriched vesicles were isolated from A2780/AD cells, non-infected Sf21 insect cells, and Sf21 cells infected with recombinant baculoviruses encoding human MDR1, rat Mdr1b, or rat Mdr2. Proteins (10 μg for A2780/AD vesicles, 2.5 μg for Sf vesicles) were separated by SDS-PAGE and transferred to a nitrocellulose filter. Immunoblotting analysis was performed using the primary antibody C219, a monoclonal antibody recognising all Pgps (Georges et al., 1990). Bound antibody was visualized as described in Methods. Lane 1: A2780/AD membrane vesicles, lane 2: Sf-MDR1 membrane vesicles, lane 3: Sf21 (non-infected) membrane vesicles, lane 4: Sf-Mdr1b membrane vesicles, lane 5: Sf-Mdr2 membrane vesicles. Molecular masses of the protein standards are indicated in kilodaltons.
ATP-dependent uptake of cationic drugs in membrane vesicles from MDR1-, Mdr1b- and Mdr2-overexpressing insect cells
In this study we used the monoquaternary quinidine and quinine derivatives N-methylquinidine and N-methylquinine as cationic model compounds (van montfoort et al., 1999), whereas the transport properties of vincristine, a classic class 1 Pgp substrate (Renes et al., 1999; Gottesman & Pastan, 1993), were used as a reference. The relative lipophilicity values of the substrates, expressed as logarithm of the octanol – TS buffer (pH 7.4) phase distribution coefficients (log D7.4 values) were 0.31±0.04, 0.11±0.02, and 1.92±0.03 (n=4) for N-methylquinidine, N-methylquinine, and vincristine, respectively.
At the beginning of our experiments we evaluated whether the three P-glycoproteins were able to transport the permanently charged cations. Figure 2A shows the ATP-dependent uptake of 200 nM [3H]-N-methylquinidine into Sf-MDR1, Sf-Mdr1b, Sf-Mdr2, and Sf21 membrane vesicles. Each data point represents a triplicate determination of a typical experiment. ATP-dependent uptake of [3H]-N-methylquinidine was observed only into Sf-MDR1 and Sf-Mdr1b membrane vesicles, whereas no ATP-dependent transport was observed into Sf21 or Sf-Mdr2 vesicles. No ATP-dependent transport was measured in membrane preparations from mock (β-glucuronidase)-infected (data not shown) and non-infected Sf21 cells. The uptake of 200 nM [3H]-N-methylquinidine into Sf-MDR1 and Sf-Mdr1b vesicles amounted to 18.01±1.77 and 8.57±0.55 pmol mg total protein−1 after 90 s for Sf-MDR1 and Sf-Mdr1b membrane vesicles, respectively.
Figure 2.
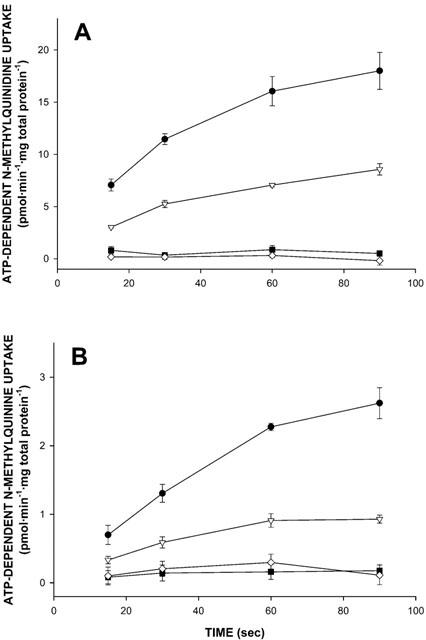
Time profiles for ATP-dependent uptake of N-methylquinidine and N-methylquinine into membrane vesicle preparations isolated from MDR1-, Mdr1b-, or Mdr2-overexpressing, or non-infected insect cells. At the time points indicated, transport of radiolabelled substrates into Sf-MDR1 (closed circles), Sf-Mdr1b (open triangles), Sf-Mdr2 (closed squares), or Sf21 (open diamonds) membrane vesicles (25 – 30 μg of protein) was determined as described in Methods. Uptake was determined for [3H]-N-methylquinidine (A), and [3H]-N-methylquinine (B), each at a final concentration of 200 nM. The uptake experiments presented in A and B were performed under identical conditions, i.e. on the same day using the same membrane vesicle preparations and reaction buffers. ATP-dependent uptake was calculated by subtracting values obtained in the presence of 4 mM AMP – PCP from those obtained in the presence of 4 mM ATP. Data points represent the mean±standard deviation of triplicate determinations of a typical experiment.
In Figure 2B the ATP-dependent uptake of 200 nM [3H]-N-methylquinine into Sf-MDR1, Sf-Mdr1b, Sf-Mdr2, and Sf21 vesicles from the same membrane preparations is presented. Like for N-methylquinidine, ATP-dependent uptake of [3H]-N-methylquinine was observed only into Sf-MDR1 and Sf-Mdr1b membrane vesicles, as compared to Sf21 vesicles, whereas no ATP-dependent transport was observed into Sf-Mdr2 vesicles. The uptake of 200 nM [3H]-N-methylquinine amounted to 2.62±0.22 and 0.93±0.06 pmol mg total protein−1 after 90 sec for Sf-MDR1 and Sf-Mdr1b membranes, respectively. As expected, ATP-dependent uptake of vincristine was only observed into Sf-MDR1 and Sf-Mdr1b membranes (data not shown).
To confirm that vesicle-associated increase of radioactivity reflects transport of the substrate into a vesicular space rather than non-specific binding to membranes, the medium osmolarity dependence of 8 μM [3H]-N-methylquinidine uptake into Sf-MDR1 and Sf-Mdr1b membrane vesicles was studied. By increasing the extravesicular sucrose concentration from 250 mM (isotonic condition) to 850 mM, the intravesicular volume decreases due to the osmolarity-mediated shrinkage of the vesicles, resulting in decreased uptake. As shown in Figure 3, initial rates of [3H]-N-methylquinidine uptake in Sf-MDR1 and Sf-Mdr1b membrane vesicles decreased linearly with increasing concentrations of sucrose. This demonstrates that increased vesicle-associated radioactivity is due to ATP-dependent uptake of the cationic substrate.
Figure 3.
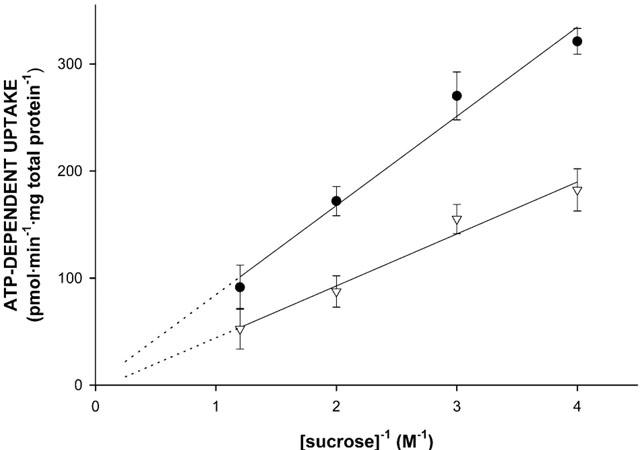
Osmolarity dependence of N-methylquinidine transport into membrane vesicle preparations isolated from MDR1- or Mdr1b-overexpressing insect cells. Sf-MDR1 (closed circles) or Sf-Mdr1b (open triangles) membrane vesicles (25 μg of protein) were incubated for 25 s at 37°C sec in the presence of 8 μM [3H]-N-methylquinidine, and different concentrations of sucrose (0.25, 0.33, 0.5 and 0.8 M). Transport of [3H]-N-methylquinidine was determined as described in Methods. ATP-dependent uptake was calculated by subtracting values obtained in the presence of 4 mM AMP – PCP from those obtained in the presence of 4 mM ATP. Data points represent the mean±standard deviation of triplicate determinations.
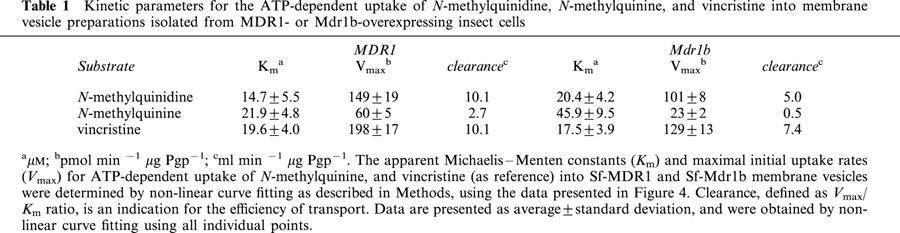
Since the same vesicle preparations were used, the data presented in Figure 2A,B suggest that N-methylquinidine is a better substrate than N-methylquinine for both MDR1 and Mdr1b. This may imply that MDR1 and Mdr1b display stereoselectivity. We therefore determined the apparent Michaelis – Menten constants, Km, and the maximal initial uptake rates, Vmax, for the uptake of these enantiomers into Sf-MDR1 and Sf-Mdr1b membrane vesicles. These two constants can be related to another parameter. The clearance of a transport protein is defined as Vmax/Km ratio. At low substrate concentrations ([substrate] < Km), clearance equals the initial rate of transport, and it is an indication for the transport capacity of a transport protein. For comparison we also determined the kinetic constants for the uptake of vincristine for both P-glycoproteins. For these experiments an uptake period of 15, 25, and 50 s was chosen to approximate initial rates of transport for vincristine, N-methylquinidine, and N-methylquinine, respectively. Since one of the aims of this study was to compare the kinetic properties of MDR1 and Mdr1b, the initial rates of transport were normalized to Pgp content of vesicle preparations and are expressed in pmol substrate min−1 μg Pgp−1. MDR1- and Mdr1b-mediated initial uptake rates of substrates were saturable and are depicted in Figure 4; the kinetic constants for the uptake are summarized in Table 1. The data points in Figure 4 represent the normalized average uptake±s.e.m. into vesicles of three different membrane preparations (two different preparations for vincristine); the uptake into each vesicle preparation was determined three times and normalized for Pgp content. All individual data points were included in the non-linear curve fitting. The apparent Km constants for ATP-dependent uptake of N-methylquinidine and N-methylquinine by MDR1 differed slightly, whereas the Km constant for Mdr1b-mediated uptake of N-methylquinine was about two-times higher than that of N-methylquinidine. For MDR1, as well as for Mdr1b, we observed that the Vmax constants for the uptake of N-methylquinine were considerably smaller than those for N-methylquinidine uptake. The clearance of the enantiomers differed 3 – 4 fold for MDR1-, and about 10 fold for Mdr1b-mediated transport. The uptake of vincristine by Sf-MDR1 or Sf-Mdr1b vesicles was half-saturated at almost equal concentrations. These concentrations are similar to that reported by Tamai & Safa (1990) for the low affinity site of MDR1. The maximal initial velocity of uptake of vincristine by MDR1 exceeded that of Mdr1b. Based on these data we conclude that N-methylquinidine is a better substrate for MDR1 and Mdr1b than N-methylquinine. In addition, our data indicate that human MDR1 has a higher transport activity than its rat orthologue Mdr1b.
Figure 4.
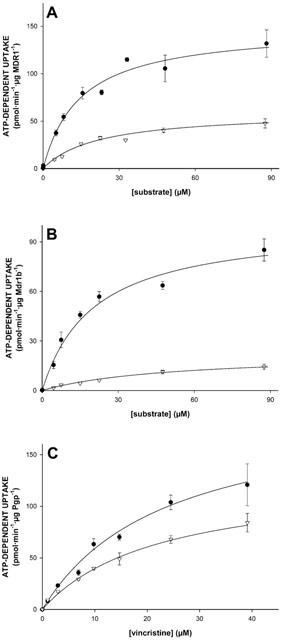
Uptake kinetics of N-methylquinidine, N-methylquinine, and vincristine by membrane vesicle preparations isolated from MDR1- or Mdr1b-overexpressing insect cells. Sf-MDR1 or Sf-Mdr1b membrane vesicles (25 μg of protein) were incubated in the presence of increasing concentrations of [3H]-labelled substrates. Initial uptake rates for N-methylquinidine, N-methylquinine, and vincristine were determined after an uptake period of 25, 50 and 15 s, respectively. Data presented are converted into uptake per min, and are corrected for Pgp-content of vesicle preparations. (A) Uptake of N-methylquinidine (closed circles) and N-methylquinine (open triangles) into Sf-MDR1 vesicles; (B) uptake of N-methylquinidine (closed circles) and N-methylquinine (open triangles) into Sf-Mdr1b vesicles; (C) uptake of vincristine into Sf-MDR1 (closed circles) and Sf-Mdr1b (open triangles) vesicles. Data points were analysed by non-linear curve fitting to an equation describing Michaelis – Menten kinetics using SigmaPlot. ATP-dependent uptake was calculated by subtracting values obtained in the presence of 4 mM AMP – PCP from those obtained in the presence of 4 mM ATP. Data points represent the normalized average uptake±standard error of the substrates into vesicles of three different membrane preparations (two for vincristine); the uptake into each vesicle preparation was determined three times and normalized for Pgp content. All individual data points were included in the non-linear curve fitting.
Table 1.
Kinetic parameters for the ATP-dependent uptake of N-methylquinidine, N-methylquinine, and vincristine into membrane vesicle preparations isolated from MDR1- or Mdr1b-overexpressing insect cells
Inhibition of [3H]-N-methylquinidine transport mediated by MDR1 and Mdr1b
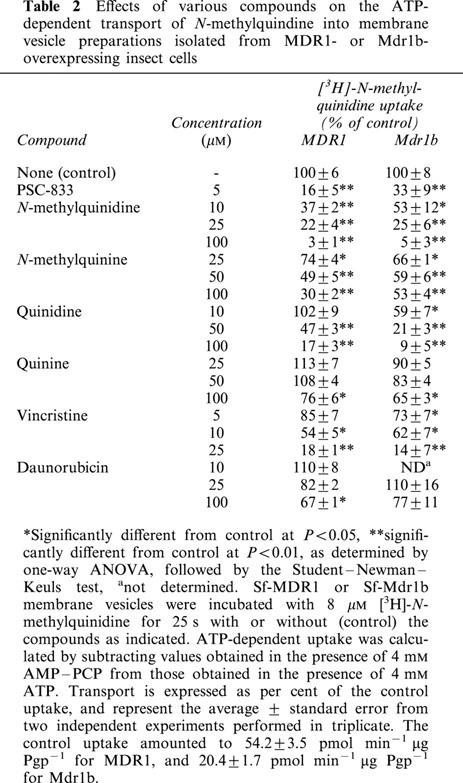
ATP-dependent [3H]-N-methylquinidine transport into Sf-MDR1 and Sf-Mdr1b membrane vesicles was measured in the presence of various potential competing substrates and inhibitors (Table 2). PSC-833, a non-immunosuppressive analogue of cyclosporin A and a potent inhibitor of Pgps (Boesch et al., 1991), profoundly inhibited the ATP-dependent uptake of 8 μM [3H]-N-methylquinidine into both Sf-MDR1 and Sf-Mdr1b membrane vesicles. An increasing concentration of unlabelled N-methylquinidine decreased the uptake of radiolabelled substrate up to almost 100%. Its diastereomer N-methylquinine also inhibited uptake of radiolabelled substrate into membrane vesicles, although less pronounced than N-methylquinidine. A similar tendency was observed for the non-methylated, weakly basic drugs quinidine and quinine. At equal concentrations quinidine was a better inhibitor of [3H]-N-methylquinidine transport than the diastereomer quinine. Uptake of [3H]-N-methylquinidine into Sf-MDR1 and Sf-Mdr1b membrane vesicles was also inhibited by the vinca alkaloid vincristine, whereas the anthracycline daunorubicin only affected the uptake of N-methylquinidine at relatively high concentrations.
Table 2.
Effects of various compounds on the ATP-dependent transport of N-methylquindine into membrane vesicle preparations isolated from MDR1- or Mdr1b- overexpressing insect cells
Discussion
Substrates for class 1 Pgps differ in their chemical structure and physicochemical properties (Ambudkar et al., 1999). Many substrates contain basic tertiary amine groups that may be protonated, depending on their pKa value and the pH of the solution, and are therefore distinct from permanently-charged cations.
In this study we have examined whether monoquaternary derivatives of the enantiomers quinidine and quinine are substrates for human MDR1, rat Mdr1b, and rat Mdr2. Compared with non- or mock-infected cells, membrane vesicles prepared from MDR1- or Mdr1b-overexpressing cells clearly showed ATP-dependent transport of these substrates. Class 1 Pgp-mediated ATP-dependent transport of the cationic substrates into membrane vesicles was saturable, occurred into an osmotically sensitive space, and could be dose-dependently inhibited by unlabelled substrate, the Pgp-inhibitor PSC-833, and various other drugs. In contrast, no Mdr2-mediated transport of the tested substrates was observed. This is in accordance with both the restricted substrate specificity and very low drug-transport capacity of Mdr2 and its human orthologue MDR3 (Buschman & Gros, 1994; Oude Elferink et al., 1997; Smith et al., 2000). Taken together, the present studies unequivocally identify the diastereomers N-methylquinidine and N-methylquinine as substrates for MDR1 and Mdr1b, but not for Mdr2.
MDR1 and Mdr1b display stereospecificity for the translocation of the two diastereomers, as is indicated by the differences in clearance, and the differential inhibition of N-methylquinidine uptake by two pairs of stereoisomers. Our data demonstrate that N-methylquinidine is a better substrate for both MDR1 and Mdr1b than its antipode N-methylquinine. Stereoselectivity for MDR1 has been noticed previously: based on the capacity of various agents to reverse resistance in Pgp-overexpressing mdr cell lines, the (R)-diastereomer quinidine has been recognized as a more potent inhibitor of Pgp than the (S)-diastereomer quinine (Akiyama et al., 1988; Genne et al., 1992). Stereoselective inhibition of MDR1 has also been reported for the diastereomers of cinchonidine (Genne et al., 1992), propranolol (Wigler & Patterson, 1994), and flupentixol (Ford et al., 1989; 1990). More recently, Dey et al. (1999) studied in more detail the interactions of the cis- and trans-isomers of flupentixol with MDR1. They showed that a common, modulatory site of MDR1 recognizes the tricyclic core of both isomers. The stereospecific inhibitory properties of these enantiomers rather result from interactions of the piperazinyl side chain with distinct regions of MDR1 near the modulatory site, that are influenced by its spatial orientation (Dey et al., 1999). However, it should be noted that in all of the above-mentioned studies Pgp-mediated transport of the compounds was not directly assayed.
The data in Table 2 indicate relatively high concentrations of quinidine, quinine, vincristine and daunorubicin are required to obtain significant inhibition of the transport of [3H]-N-methylquinidine. Moreover, at even high concentrations (100 μM) the classic Pgp substrate daunorubicin inhibited the transport only slightly (23 – 33%). This suggests that N-methylquinidine binds to Pgp at a site that is distinct from the site of interaction of daunorubicin, being in line with recent studies that indicate Pgp contains multiple drug binding sites rather than a single site of broad specificity (Martin et al., 2000; Dey et al., 1999; Garrigos et al., 1997; Ambudkar et al., 1999).
Presently, a definite molecular explanation for the observed stereoselective transport cannot be given. The wide variety of data on the potential mechanisms of class 1. Pgp-mediated transport indicates at least two transport modalities (discussed in more detail below). Class 1 Pgps may extract amphipathic substrates from the membrane releasing it into the extracellular aqueous phase, or, alternatively, class 1 Pgps may operate as a ‘pump', translocating substrates across the membrane directly from the cytosol into the extracellular fluid. In the current study we directly assayed the MDR1- and Mdr1b-mediated transport of N-methylquinidine and N-methylquinine. Figure 2A,B show that under identical conditions, i.e. equal concentrations of substrates and MDR1 or Mdr1b, the initial uptake of N-methylquinidine is greater than of N-methylquinine for both MDR1 and Mdr1b. In contrast to the marked differences between the normalized Vmax constants for the MDR1- and Mdr1b-mediated uptake of the enantiomers, we found that the apparent Km constants differ only slightly. This indicates that the differences in handling of the isomers by MDR1 and Mdr1b are due to variations in substrate handling at a point subsequent to the initial binding interaction. Moreover, this may imply that post-binding dissociation events, but not binding itself, represents the rate-limiting step in Pgp-mediated transport of these enantiomers.
We also provide evidence that human MDR1 has a higher transport activity than its rat orthologue Mdr1b, as is indicated by the differences in clearance. Despite their high sequence similarity (80% at amino acid level), the observation that functional differences exist is not unexpected. It has been shown that the two murine drug transporting isoforms, Mdr1a and Mdr1b, display overlapping, but distinct drug resistance phenotypes (Tang-Wai et al., 1995), and transport [3H]-vinblastine with different efficiencies (Yang et al., 1990), in spite of their equal affinity and binding capacity towards this substrate (Taylor et al., 1999). Moreover, as summarized by Ambudkar et al. (1999), studies with mutated Pgps revealed that a change of only one amino acid may already dramatically affect the initial rate of drug uptake in accumulation assays with transfected cells. Nevertheless, the observation that functional differences exist between human MDR1 and rat Mdr1b may be of interest not only from a biochemical, but also from a pharmacological perspective. Since drug transporting Pgp isoforms play an important role the absorption, distribution, and elimination of drugs, detailed knowledge of the functional properties of each individual isoform may eventually result into extrapolation to humans of data obtained in rats with regard to bioavailability and safety of Pgp substrates.
As previously stated, the molecular mechanism by which class 1 Pgps perform their transport function is still a subject of debate. Class 1 Pgps may both reduce the influx rate of drugs into the cytoplasm, and increase the efflux out of multidrug resistant cells (Gottesman & Pastan, 1993; Stein et al., 1994). It has been hypothesized that class 1 Pgps recognize and remove hydrophobic drugs directly from the lipid bilayer, for example in a process comparable to the action of a hydrophobic vacuum cleaner (Raviv et al., 1990). Another version of this model suggests that class 1 Pgps act as flippases (Higgins & Gottesman, 1992) that translocate drugs from the inner leaflet to the outer leaflet of the membrane, where it would passively equilibrate with the aqueous medium. Stein (1997) proposes that substrates are moved into a ‘medium-accessible' cavity, formed by the class 1 Pgp molecule, before they are released into the external medium. Interestingly, Altenberg et al. (1994) present an opposite working model. These authors speculate that class 1 Pgps operate as an efflux pump, transporting substrates directly from the cytosol into the extracellular medium. The results of this paper show that N-methylquinidine is an excellent substrate for both MDR1 and Mdr1b, with a transport capacity that comes close to that of the much more hydrophobic drug vincristine. In view of the marked differences in log D7.4 values it is unlikely that N-methylquinidine and N-methylquinine will partition into the lipid phase of membranes to the same extent as vincristine. In this respect, the presently studied N-alkylated derivatives of quinidine and quinine, that have a permanently positively charged nitrogen centre, may behave fundamentally different from the tertiary parent compounds. The latter are able to diffuse into, and to cross membranes in the non-charged lipophilic form. Consequently, our observations do not fit well in the mentioned models that assume class 1 Pgps extract its substrate from the lipid bilayer. Our data are more comparable with models that postulate substrates are removed directly from the cytosol. Yet, this leaves open the possibility that the monoquaternary cations studied can enter a putative ‘active site' from the lipid bilayer.
In conclusion, we have demonstrated that permanently positively charged derivatives of the diastereomers quinidine and quinine are transported by human MDR1 and rat Mdr1b, but not by rat Mdr2. Moreover, the (R)-diastereomer N-methylquinidine is a better substrate for both MDR1 and Mdr1b than the (S)-diastereomer N-methylquinine. Finally, our data indicate human MDR1 is more efficient in transporting drugs than its rat orthologue Mdr1b.
Acknowledgments
The authors thank Hans Koning (Div. Gastroenterology and Hepatology) for excellent technical assistance, Dr Douwe F. Westra (Dept. Medical Microbiology, University of Groningen) for assistance in developing the baculovirus expression system, Dr Jeffrey A. Silverman (AvMax Inc., Berkeley, U.S.A.) for providing the rat Mdr1b and Mdr2 cDNA constructs, and Dr U.S. Rao (University of North Carolina, Chapel Hill, U.S.A.) for providing an aliquot of a high titer baculovirus stock containing the human MDR1 gene. This work was supported by Netherlands Organization for Scientific Research (NWO) grant NWO 902-23-207 (D.K.F. Meijer, M. Müller). J.E. v Montfoort was a recipient of an Ubbo Emmius scholarship of the University of Groningen.
Abbreviations
- Mdr
multidrug resistance
- MDR
Mdr, genes encoding human or rodent multidrug resistance proteins, respectively
- MDR
Mdr, human or rodent multidrug resistance proteins, respectively
- Pgp
P-glycoprotein
- Sf
Spodoptera frugiperda
References
- AKIYAMA S., CORNWELL M.M., KUWANO M., PASTAN I., GOTTESMAN M.M. Most drugs that reverse multidrug resistance also inhibit photoaffinity labeling of P-glycoprotein by a vinblastine analog. Mol. Pharmacol. 1988;33:144–147. [PubMed] [Google Scholar]
- ALTENBERG G.A., VANOYE C.G., HORTON J.K., REUSS L. Unidirectional fluxes of rhodamine 123 in multidrug-resistant cells: evidence against direct drug extrusion from the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4654–4657. doi: 10.1073/pnas.91.11.4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMBUDKAR S.V., DEY S., HRYCYNA C.A., RAMACHANDRA M., PASTAN I., GOTTESMAN M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- BOESCH D., GAVERIAUX C., JACHEZ B., POURTIER-MANZANEDO A., BOLLINGER P., LOOR F. In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res. 1991;51:4226–4233. [PubMed] [Google Scholar]
- BROWN P.C., THORGEIRSSON S.S., SILVERMAN J.A. Cloning and regulation of the rat mdr2 gene. Nucleic. Acids. Res. 1993;21:3885–3891. doi: 10.1093/nar/21.16.3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSCHMAN E., GROS P. The inability of the mouse mdr2 gene to confer multidrug resistance is linked to reduced drug binding to the protein. Cancer Res. 1994;54:4892–4898. [PubMed] [Google Scholar]
- CHEN C.J., CHIN J.E., UEDA K., CLARK D.P., PASTAN I., GOTTESMAN M.M., RONINSON I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- DEUCHARS K.L., DUTHIE M., LING V. Identification of distinct P-glycoprotein gene sequences in rat. Biochim. Biophys. Acta. 1992;1130:157–165. doi: 10.1016/0167-4781(92)90523-3. [DOI] [PubMed] [Google Scholar]
- DEVAULT A., GROS P. Two members of the mouse mdr gene family confer multidrug resistance with overlapping but distinct drug specificities. Mol. Cell Biol. 1990;10:1652–1663. doi: 10.1128/mcb.10.4.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEY S., HAFKEMEYER P., PASTAN I., GOTTESMAN M.M. A single amino acid residue contributes to distinct mechanisms of inhibition of the human multidrug transporter by stereoisomers of the dopamine receptor antagonist flupentixol. Biochemistry. 1999;38:6630–6639. doi: 10.1021/bi983038l. [DOI] [PubMed] [Google Scholar]
- FORD J.M., BRUGGEMANN E.P., PASTAN I., GOTTESMAN M.M., HAIT W.N. Cellular and biochemical characterization of thioxanthenes for reversal of multidrug resistance in human and murine cell lines. Cancer Res. 1990;50:1748–1756. [PubMed] [Google Scholar]
- FORD J.M., PROZIALECK W.C., HAIT W.N. Structural features determining activity of phenothiazines and related drugs for inhibition of cell growth and reversal of multidrug resistance. Mol. Pharmacol. 1989;35:105–115. [PubMed] [Google Scholar]
- GARRIGOS M., MIR L.M., ORLOWSKI S. Competitive and non-competitive inhibition of the multidrug- resistance- associated P-glycoprotein ATPase-further experimental evidence for a multisite model. Eur. J. Biochem. 1997;244:664–673. doi: 10.1111/j.1432-1033.1997.00664.x. [DOI] [PubMed] [Google Scholar]
- GENNE P., DIMANCHE-BOITREL M.T., MAUVERNAY R.Y., GUTIERREZ G., DUCHAMP O., PETIT J.M., MARTIN F., CHAUFFERT B. Cinchonine, a potent efflux inhibitor to circumvent anthracycline resistance in vivo. Cancer Res. 1992;52:2797–2801. [PubMed] [Google Scholar]
- GEORGES E., BRADLEY G., GARIEPY J., LING V. Detection of P-glycoprotein isoforms by genespecific monoclonal antibodies. Proc. Natl. Acad. Sci. U.S.A. 1990;87:152–156. doi: 10.1073/pnas.87.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERMANN U.A., WILLINGHAM M.C., PASTAN I., GOTTESMAN M.M. Expression of the human multidrug transporter in insect cells by a recombinant baculovirus. Biochemistry. 1990;29:2295–2303. doi: 10.1021/bi00461a013. [DOI] [PubMed] [Google Scholar]
- GOTTESMAN M.M., PASTAN I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- HIGGINS C.F., GOTTESMAN M.M. Is the multidrug transporter a flippase. Trends. Biochem. Sci. 1992;17:18–21. doi: 10.1016/0968-0004(92)90419-a. [DOI] [PubMed] [Google Scholar]
- KAMIMOTO Y., GATMAITAN Z., HSU J., ARIAS I.M. The function of Gp170, the multidrug resistance gene product, in rat liver canalicular membrane vesicles. J. Biol. Chem. 1989;264:11693–11698. [PubMed] [Google Scholar]
- LEE C., LEVIN A., BRANTON D. Copper staining: a five-minute protein stain for sodium dodecyl sulfate-polyacrylamide gels. Anal. Biochem. 1987;166:308–312. doi: 10.1016/0003-2697(87)90579-3. [DOI] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., HIGGINS C.F., MISTRY P., CHARLTON P., CALLAGHAN R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- MÜLLER M., MAYER R., HERO U., KEPPLER D. ATP-dependent transport of amphiphilic cations across the hepatocyte canalicular membrane mediated by mdr1 P-glycoprotein. FEBS Lett. 1994a;343:168–172. doi: 10.1016/0014-5793(94)80312-9. [DOI] [PubMed] [Google Scholar]
- MÜLLER M., MEIJER C., ZAMAN G.J.R., BORST P., SCHEPER R.J., MULDER N.H., DE VRIES E.G.E., JANSEN P.L.M. Overexpression of the gene encoding the multidrug resistance- associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc. Natl. Acad. Sci. U.S.A. 1994b;91:13033–13037. doi: 10.1073/pnas.91.26.13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OUDE ELFERINK R.P.J., TYTGAT G.N.J., GROEN A.K. Hepatic canalicular membrane 1: The role of mdr2 P-glycoprotein in hepatobiliary lipid transport. FASEB J. 1997;11:19–28. doi: 10.1096/fasebj.11.1.9034162. [DOI] [PubMed] [Google Scholar]
- RAO U.S. Mutation of glycine 185 to valine alters the ATPase function of the human P-glycoprotein expressed in Sf9 cells. J. Biol. Chem. 1995;270:6686–6690. [PubMed] [Google Scholar]
- RAVIV Y., POLLARD H.B., BRUGGEMANN E.P., PASTAN I., GOTTESMAN M.M. Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J. Biol. Chem. 1990;265:3975–3980. [PubMed] [Google Scholar]
- RENES J., DE VRIES E.G.E., NIENHUIS E.F., JANSEN P.L.M., MÜLLER M. ATP- and glutathione-dependent transport of chemotherapeutic drugs by the multidrug resistance protein MRP1. Br. J. Pharmacol. 1999;126:681–688. doi: 10.1038/sj.bjp.0702360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARKADI B., PRICE E.M., BOUCHER R.C., GERMANN U.A., SCARBOROUGH G.A. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 1992;267:4854–4858. [PubMed] [Google Scholar]
- SCHINKEL A.H. Pharmacological insights from P-glycoprotein knockout mice. Int. J. Clin. Pharmacol. Ther. 1998;36:9–13. [PubMed] [Google Scholar]
- SILVERMAN J.A., RAUNIO H., GANT T.W., THORGEIRSSON S.S. Cloning and characterization of a member of the rat multidrug resistance (mdr) gene family. Gene. 1991;106:229–236. doi: 10.1016/0378-1119(91)90203-n. [DOI] [PubMed] [Google Scholar]
- SMIT J.W., SCHINKEL A.H., MÜLLER M., WEERT B., MEIJER D.K.F. Contribution of the murine mdr1a P-glycoprotein to hepatobiliary and intestinal elimination of cationic drugs as measured in mice with an mdr1a gene disruption. Hepatology. 1998a;27:1056–1063. doi: 10.1002/hep.510270422. [DOI] [PubMed] [Google Scholar]
- SMIT J.W., SCHINKEL A.H., WEERT B., MEIJER D.K.F. Hepatobiliary and intestinal clearance of amphiphilic cationic drugs in mice in which both mdr1a and mdr1b genes have been disrupted. Br. J. Pharmacol. 1998b;124:416–424. doi: 10.1038/sj.bjp.0701845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMIT J.W., WEERT B., SCHINKEL A.H., MEIJER D.K.F. Heterologous expression of various P-glycoproteins in polarized epithelial cells induces directional transport of small (type 1) and bulky (type 2) cationic drugs. J. Pharmacol. Exp. Ther. 1998c;286:321–327. [PubMed] [Google Scholar]
- SMITH A.J., VAN HELVOORT A., VAN MEER G., SZABO K., WELKER E., SZAKACS G., VARADI A., SARKADI B., BORST P. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J. Biol. Chem. 2000;275:23530–23539. doi: 10.1074/jbc.M909002199. [DOI] [PubMed] [Google Scholar]
- STEIN W.D. Kinetics of the multidrug transporter (P-glycoprotein) and its reversal. Physiol. Rev. 1997;77:545–590. doi: 10.1152/physrev.1997.77.2.545. [DOI] [PubMed] [Google Scholar]
- STEIN W.D., CARDARELLI C., PASTAN I., GOTTESMAN M.M. Kinetic evidence suggesting that the multidrug transporter differentially handles influx and efflux of its substrates. Mol. Pharmacol. 1994;45:763–772. [PubMed] [Google Scholar]
- TAMAI I., SAFA A.R. Competitive interaction of cyclosporins with the Vinca alkaloid-binding site of P-glycoprotein in multidrug-resistant cells. J. Biol. Chem. 1990;265:16509–16513. [PubMed] [Google Scholar]
- TANG-WAI D.F., KAJIJI S., DICAPUA F., DE GRAAF D., RONINSON I.B., GROS P. Human (MDR1) and mouse (mdr1, mdr3) P-glycoproteins can be distinguished by their respective drug resistance profiles and sensitivity to modulators. Biochemistry. 1995;34:32–39. doi: 10.1021/bi00001a005. [DOI] [PubMed] [Google Scholar]
- TAYLOR J.C., FERRY D.R., HIGGINS C.F., CALLAGHAN R. The equilibrium and kinetic drug binding properties of the mouse P-gp1a and P-gp1b P-glycoproteins are similar. Br. J. Cancer. 1999;81:783–789. doi: 10.1038/sj.bjc.6690764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THIEBAUT F., TSURUO T., HAMADA H., GOTTESMAN M.M., PASTAN I., WILLINGHAM M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN MONTFOORT J.E., HAGENBUCH B., FATTINGER K., MÜLLER M., GROOTHUIS G.M.M., MEIJER D.K.F., MEIER P.J. Polyspecific organic anion transporting polypeptides mediate hepatic uptake of amphipatic type II organic cations. J. Pharmacol. Exp. Ther. 1999;291:147–152. [PubMed] [Google Scholar]
- WATANABE T., MIYAUCHI S., SAWADA Y., IGA T., HANANO M., INABA M., SUGIYAMA Y. Kinetic analysis of hepatobiliary transport of vincristine in perfused rat liver. Possible roles of P-glycoprotein in biliary excretion of vincristine. J. Hepatol. 1992;16:77–88. doi: 10.1016/s0168-8278(05)80098-4. [DOI] [PubMed] [Google Scholar]
- WIGLER P.W., PATTERSON F.K. Reversal agent inhibition of the multidrug resistance pump in human leukemic lymphoblasts. Biochim. Biophys. Acta. 1994;1189:1–6. doi: 10.1016/0005-2736(94)90272-0. [DOI] [PubMed] [Google Scholar]
- YANG C.P.H., COHEN D., GREENBERGER L.M., HSU S.I.H., HORWITZ S.B. Differential transport properties of two mdr gene products are distinguished by progesterone. J. Biol. Chem. 1990;265:10282–10288. [PubMed] [Google Scholar]