

Abstract
We have previously shown that the flavonoid luteolin inhibits the expression of pro-inflammatory molecules induced by LPS. In the present study we tested the ability of luteolin to block signalling pathways implicated in LPS-induced inflammatory gene expression in macrophages.
Exposure of the murine macrophage cell line RAW 264.7 to LPS increased phosphorylation of the mitogen-activated protein kinase family members ERK1/2, p38 and JNK1/2 in a time-dependent manner. Pretreatment of RAW 264.7 with luteolin inhibited the LPS-induced ERK1/2 and p38, but not JNK1/2, phosphorylation, and blocked the LPS-induced TNF-α release.
To investigate which of these pathways contribute to the inhibitory effects of luteolin on TNF-α release, cells were pretreated with pharmacological inhibitors of these pathways; PD98059 and SB203580 when used alone failed to inhibit TNF-α release, whereas pretreatment with both agents attenuated TNF-α release.
We have previously shown that luteolin blocks Akt phosphorylation in response to LPS in RAW 264.7 macrophages. To determine the role of Akt in TNF-α release, cells were transiently transfected with a dominant negative form of Akt (K179M). Overexpression of K179M Akt did not alter LPS-induced TNF-α release, suggesting that inhibition of this kinase does not mediate the inhibitory action of luteolin.
In addition, DRB (a pharmacological inhibitor of CK2) blocked TNF-α release in a concentration-dependent manner, whereas co-treatment of cells with luteolin and DRB did not have an additive effect.
We conclude that luteolin interferes with LPS signalling by reducing the activation of several MAPK family members and that its inhibitory action on TNF-α release correlates with inhibition of ERK, p38 and CK2 activation.
Keywords: Tumour necrosis factor-α, lipopolysaccharide, luteolin, casein kinase 2, MAPK
Introduction
Lipopolysaccharide (LPS) is a component of the cell wall of Gram-negative bacteria that binds to mammalian cell membrane molecules such as CD14 and toll-like receptors (TLR) (Hoshino et al., 1999; Wright et al., 1990). Among the ten members of the TLR family reported so far, TLR-4 and TLR-2 play an important role in the LPS response; TLR-4 is thought to be the principal LPS receptor, whereas TLR-2 recognizes primarily lipoproteins and glycolipids that are often found in commercially available LPS preparations (Akira et al., 2001). Binding of LPS to its cognate receptors activates several signalling cascades driving the expression of pro-inflammatory cytokines and adhesion molecules that contribute to the pathogenesis of septic shock (Karima et al., 1999). One cytokine that plays a key role in sepsis is tumour necrosis factor α (TNF-α) (Vassalli, 1992). Various therapeutic strategies, including administration of neutralizing antibodies and soluble receptors for TNF-α, as well as pharmacological agents that block its production have been employed in animal models of sepsis improving survival (Baumgartner & Calandra, 1999; Novogrodsky et al., 1994).
Macrophages are an important source of TNF-α during sepsis and a number of pathways have been suggested to contribute to the LPS-induced TNF-α production in this cell type (Geppert et al., 1994; Kontoyiannis et al., 1999; Novogrodsky et al., 1994). Following exposure to LPS, several proteins become tyrosine phosphorylated and broad-spectrum tyrosine kinase inhibitors, such as the tyrphostins, block LPS-induced activation of downstream kinases and TNF-α release (Novogrodsky et al., 1994). A group of downstream ser/thr kinases that become activated in response to LPS regulating TNF-α release in macrophages are the mitogen-activated protein kinase (MAPK) family members (Chang & Karin, 2001). Many studies have demonstrated that inhibition of extracellular signal-regulated (ERK) 1/2 and p38 activation affects LPS-stimulated TNF-α production from macrophages, although differences have been observed between cell lines and cells isolated from different tissues (Baldassare et al., 1999; Carter et al., 1999; Feng et al., 1999; Means et al., 2000; van-den-Blink et al., 2001). Activation of the different MAPK signalling cascades is believed to control different steps in the TNF-α production process. Activation of ERK1/2 has been reported to stimulate TNF-α transcription and control the transport of TNF-α mRNA from the nucleus to the cytoplasm (Dumitru et al., 2000; Geppert et al., 1994). On the other hand, p38 and JNK control TNF-α mRNA stability and TNF-α translation (Kontoyiannis et al., 1999; Swantek et al., 1997).
Flavonoids are naturally occurring compounds with anti-inflammatory properties (Middleton et al., 2001). Others and we, have previously reported that flavonoids inhibit LPS-induced expression of TNF-α, interleukin-6, nitric oxide and intercellular adhesion molecule-1 (Gerritsen et al., 1995; Wadsworth & Koop, 1999; Xagorari et al., 2001). In a previous study we identified luteolin as the most potent and efficacious of the flavonoids tested in inhibiting TNF-α release in murine macrophages, and showed that luteolin blocks LPS-induced activation of nuclear factor κB (NF-κB), as well as NF-κB-driven gene expression (Xagorari et al., 2001). Moreover, we have recently demonstrated that luteolin increases survival in an animal model of sepsis (Kotanidou et al., 2002). The aim of the present study was to identify additional molecular targets for luteolin and correlate inhibition of specific signalling molecules with the inhibitory action on TNF-α release.
Methods
Reagents
Luteolin and myricetin were obtained from Roth Chemicalien (Karlsruhe, Germany). TNF-α ELISA kits were from R&D Systems (Minneapolis, MN, U.S.A.). Tissue culture plates were from Greiner GmbH (Germany). Dulbecco's modified Eagle's medium (DMEM), foetal calf serum, antibiotics, and LipofectAmine were obtained from Life Technologies (Paisley, U.K.). The nitrocellulose membrane was obtained from Bio-Rad (Hercules, CA, U.S.A.) and the Supersignal Chemiluminescent Substrate from Pierce (Rockford, K, U.S.A.). The pharmacological inhibitors SB203580, PD98059, LY294002, wortmannin and DRB were purchased by Calbiochem-Novabiochem GmbH (Germany). Rabbit polyclonal antibodies directed against the phosphorylated forms of MEK1/2, MKK3/6, ERK1/2, p38 and JNK as well as the total forms of the above-mentioned kinases were purchased from Cell Signalling (Beverly, MA, U.S.A.). Anti-rabbit IgG (Goat), HRP-labelled antibody was purchased from NEN Life Science Products, Inc (Boston, MA, U.S.A.). Reagents for SDS-polyacrylamide gel electrophoresis were obtained from Biorad (Hercules, CA, U.S.A.). All other reagents including LPS (E. coli 026 : B6) were obtained from Sigma Chemical Co. (St Louis, MO, U.S.A.).
Cell culture and cytokine measurements
RAW264.7 murine macrophages were cultured in Dulbecco's MEM containing 10% foetal bovine and 2 mM L-glutamine serum supplemented with penicillin and streptomycin, at 37°C in a humidified incubator with 5% CO2. For the cytokine measurements cells were exposed to the indicated concentration of luteolin, myricetin, SB203580, PD98059, LY294002 or wortmannin for 30 min or 60 min before the LPS challenge (10 ng ml−1). After 24 h supernatants were collected and centrifuged for 10 min at 3000 r.p.m. in a tabletop micro-centrifuge to remove non-adherent cells. Following centrifugation, pellets were discarded and supernatants used for ELISA in accordance to the manufacturer's instructions.
Immunoblotting
RAW 264.7 cells were serum starved for 10 h (for the ERK1/2, JNK1/2 and p38 experiments) or 20 h (for the MEK1/2 and MKK3/6 experiments) and treated as described in the figure legends. Cells were then lysed in a buffer containing 1% NP40, 50 mM NaCl, 0.1% SDS, 50 mM NaF, 1 mM NaVO4, 50 mM Tris-HCl, 0.1 mM EGTA, 0.5% deoxycholic acid, 1 mM EDTA, 1 μg ml−1 aprotinin, 1 μg ml−1 leupeptin, 1 μg ml−1 pepstatin and 1 mM PMSF. Sample aliquots (50 μg/lane) were subjected to standard SDS–PAGE. Following antibody exposure, immunoreactive protein bands were visualized using a chemiluminescent substrate.
Transfections
RAW 264.7 cells were plated in 6 well plates at a density of 2×104 per square centimeter and allowed to reach 40–60% confluence. Cells were transfected with vector alone (pCMV–βgal) or a dominant negative form of Akt (K179M). Transfections were performed using LipofectAMINE at a DNA/lipid of 2 μg plasmid DNA/3 μl lipid; transfection efficiency was typically 65% or greater. Twenty-four hours after the initiation of transfection, cells were challenged with LPS (10 ng ml−1) for an additional 24 h; cell culture supernatants were then collected and TNF-α measured.
Data analysis and statistics
Data are presented as means±s.e.mean of the indicated number of observations. Cytokine values are expressed as ng ml−1 or as per cent of the LPS value. Statistical comparisons between groups were performed using the one-way Anova followed by the Dunnett's or Newman-Keuls post hoc test or the Student's t-test, as appropriate. Differences among means were considered significant when P<0.05.
Results
Effects of luteolin on LPS-induced MAPK phosphorylation
Exposure of RAW 264.7 to LPS (10 ng ml−1) stimulated the phosphorylation of MAPK signalling cascades in a time-dependent manner (Figure 1). Activation of ERK1/2 and p38 is an earlier event than JNK1/2 activation as it was evident within 5 min of the LPS challenge, whereas phosphorylation of JNK was more pronounced after 20 min. To determine if luteolin has the ability to block the activation of MAPKs, macrophages were pretreated with 10 μM luteolin 30 min prior to the LPS challenge. Such pretreatment resulted in the blockade of LPS-induced ERK1/2 and p38, but not JNK phosphorylation, without affecting the total levels of these kinases (Figure 2). In addition, luteolin blocked the LPS-induced MEK1/2 and MKK3/6 phosphorylation, suggesting that this flavonoid acts upstream of the MAPKKs (Figure 3).
Figure 1.
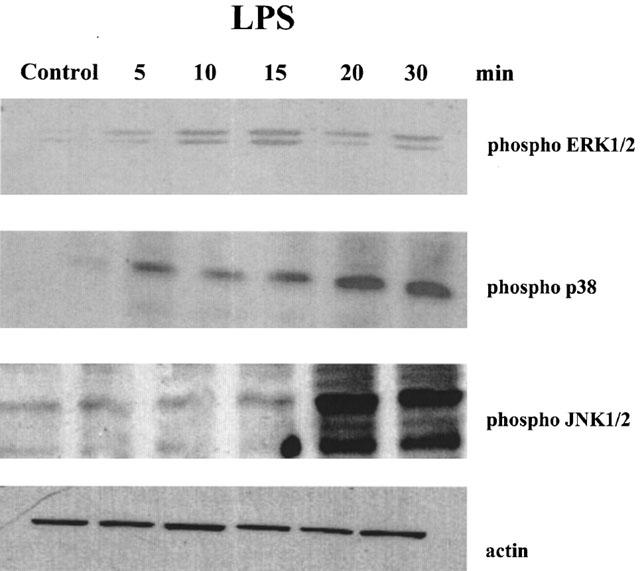
LPS induces phosphorylation of p38, ERK1/2 and JNK1/2 in a time-dependent manner. RAW 264.7 cells were incubated with LPS (10 ng ml−1) for the indicated time. Total cell lysates were processed by SDS–PAGE and membranes blotted with phosphospecific antibodies for p38, ERK1/2, JNK or an antibody that recognizes all actin isoforms. Similar results were obtained in three different experiments.
Figure 2.
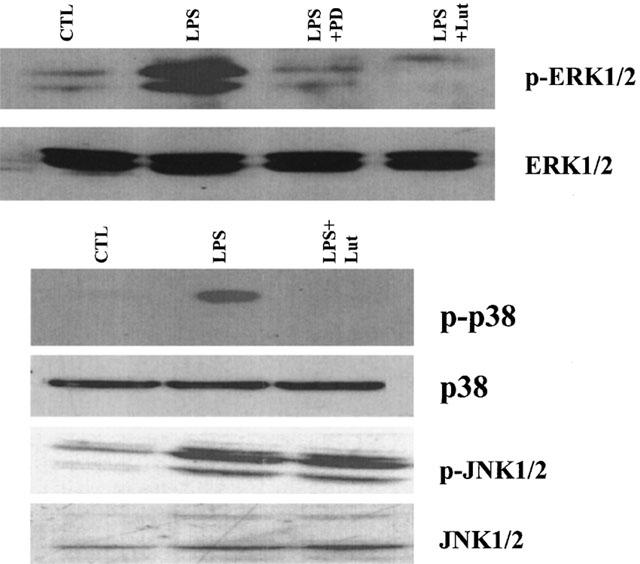
Luteolin inhibits LPS-induced ERK1/2 and p38 phosphorylation. RAW 264.7 cells were pretreated with luteolin (10 μM, 30 min) or PD98059 (10 μM, 1 h) and then exposed to LPS (10 ng ml−1) for 15 min (ERK1/2) or 20 min (p38 and JNK). Total cell lysates were processed by SDS–PAGE and membranes blotted with a phosphospecific ERK1/2, p38 or JNK1/2 antibody. Membranes run in parallel were blotted with antibodies that recognize total cellular ERK1/2, JNK1/2 or p38. Similar results were obtained in two different experiments.
Figure 3.
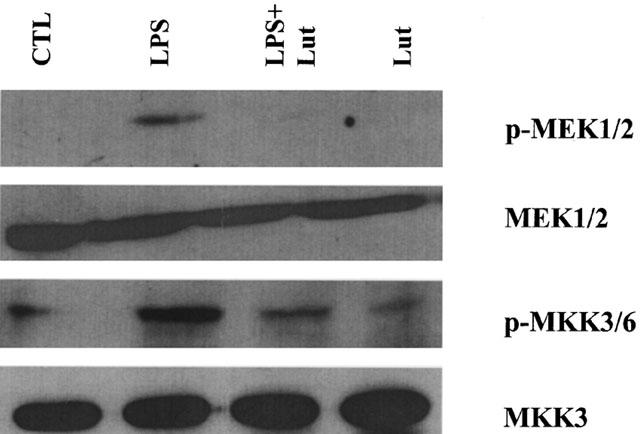
Luteolin inhibits LPS-induced MAPKK phosphorylation. RAW 264.7 cells were pretreated with luteolin (10 μM, 30 min) prior to LPS (10 ng ml−1) and MEK1/2 and MKK3/6 phosphorylation was determined after 20 min using phospho-specific antibodies. Membranes run in parallel were blotted with antibodies that recognize total cellular MEK1/2 or MKK3. Similar results were obtained in three different experiments.
Effect of inhibition of ERK and p38 pathways on LPS-induced TNF-α release
To determine if inhibition of ERK or p38 kinase contributes to the inhibitory action of luteolin, cells were pretreated with the MEK1/2 and p38 kinase inhibitors PD98059 and SB203580, respectively. Over the concentration range used (0.1–10 μM for PD98059 and 0.1–2.5 μM for SB203580) neither inhibitor had any significant effect on LPS-induced TNF-α release when used alone (Figure 4). However, pretreatment of cells with a combination of 10 μM PD98059 and 2.5 μM SB203580 blocked the LPS-response by 67.7%.
Figure 4.
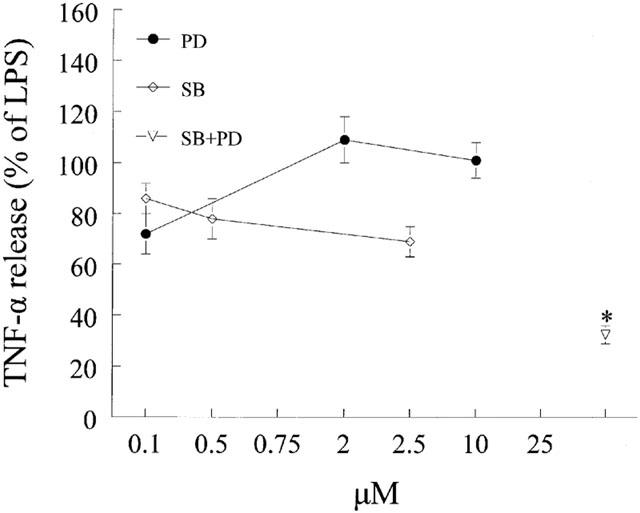
Effects of p38 and MEK1/2 inhibition on LPS-induced TNF-α release from mouse macrophages. Cells were pretreated for 1 h with vehicle (DMSO), SB203580 (SB) or PD98059 (PD) at the indicated concentration or a combination of SB plus PD (2.5 μM and 10 μM). At the end of pretreatment, macrophages were challenged with LPS (10 ng ml−1) for 24 h and media collected and processedas described in the Methods section. Data are presented as means±s.e.mean; n=11; *P<0.05 from LPS.
Effect of inhibition of the PI3-K and Akt on LPS-induced TNF-α release
Flavonoids are known to inhibit PI3-K activity (Gamet-Payrastre et al., 1999). Moreover, we have demonstrated that luteolin blocks LPS-induced Akt phopshorylation (Xagorari et al., 2001). To determine if inhibition of PI3-K contributes to the inhibitory action of luteolin on TNF-α release, we pretreated cells with two chemically distinct PI3-K inhibitors, LY294002 or wortmannin (Figure 5). Pretreatment of the cells with LY294002 resulted in significant inhibition of TNF-α release only at the highest concentration used (25 μM), whereas wortmannin was ineffective in blocking the production of this cytokine. Moreover, pretreatment of cells with myricetin, a flavonoid known to inhibit PI3-K (Gamet-Payrastre et al., 1999) did not affect LPS-induced TNF-α release, providing further evidence that inhibition of PI3-K does not lead to a reduction in TNF-α production in response to LPS (Figure 6). To characterize the role of Akt on TNF-α release, cells were transiently transfected with a dominant negative form of Akt (K179M). Overexpression of this kinase-dead form of Akt did not block the effects of LPS on TNF-α release (Figure 7), suggesting that activation of Akt does not play a major role in the production of TNF-α.
Figure 5.
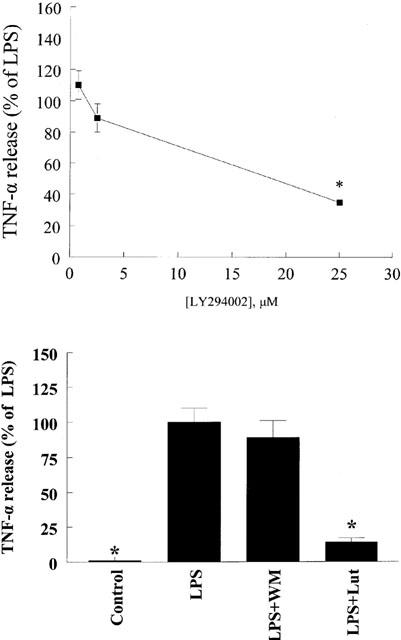
Effects of PI3-K inhibition on LPS-induced TNF-α release from mouse macrophages. Cells were pretreated with the indicated concentration of LY294002 for 1 h (top) and luteolin (Lut, 10 μM) or wortmannin (WM, 10 nM) for 30 min (bottom). At the end of pretreatment, macrophages were exposed to LPS (10 ng ml−1) for 24 h. Culture supernatants were collected and TNF-α levels determined by ELISA. Data are presented as means±s.e.mean; n=7–12; *P<0.05 from LPS.
Figure 6.
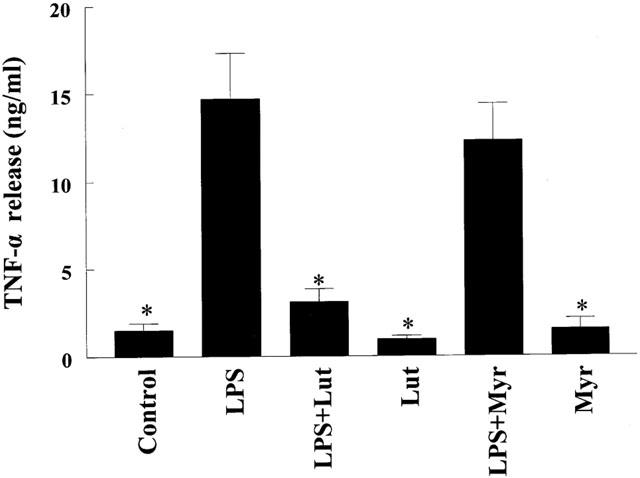
Effects of myricetin on LPS-induced TNF-α release from mouse macrophages. Cells were pretreated for 30 min with 10 μM myricetin (Myr), 10 μM luteolin (Lut), or vehicle (DMSO : EtOH; 1 : 1,v v−1). At the end of pretreatment, macrophages were incubated with LPS (10 ng ml−1) for 24 h and media collected and analysed as described in Methods. Data are presented as means±s.e.mean; n=8–12; *P<0.05 from LPS.
Figure 7.
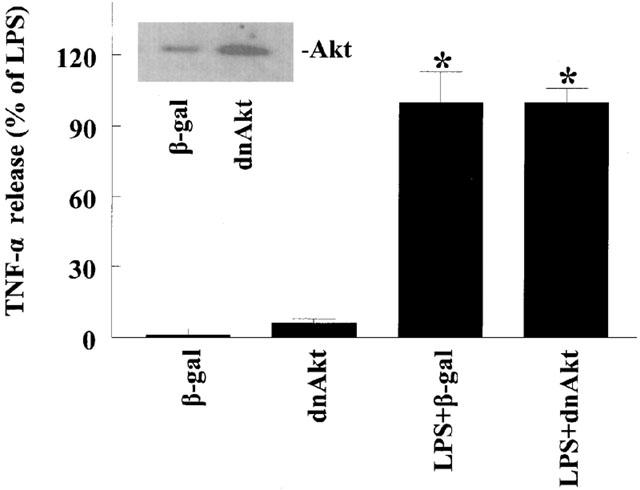
Inhibition of Akt does not affect LPS-induced TNF-α release from macrophages. RAW 264.7 cells were transfected with a dominant negative form of Akt (K179M) or a plasmid expressing β-galactosidase as control. After 24 h cells were challenged with LPS (10 ng ml−1; 24 h). Culture supernatants were collected and TNF-α levels determined by ELISA. The inset confirms expression of the dominant negative Akt. Data are presented as means±s.e.mean; n=5; *P<0.05 from respective control values.
Effect of CK2 inhibition on LPS-induced TNF-α release
Although LY294002 is considered a specific PI3-K inhibitor it can also block CK2 activity (Davies et al., 2000). To address the role of CK2 on TNF-α production, cells were pretreated with the pharmacological inhibitor of CK2 5,6-dichlororibifuranosylbenzimidasole (DRB). Pretreatment with DRB resulted in a concentration-dependent inhibition of LPS-induced TNF-α production (Figure 8). Moreover, the inhibitory effect of luteolin and DRB co-treatment on TNF-α release was not greater than that observed with luteolin alone, suggesting that part of the action of luteolin is mediated through inhibition of CK2.
Figure 8.
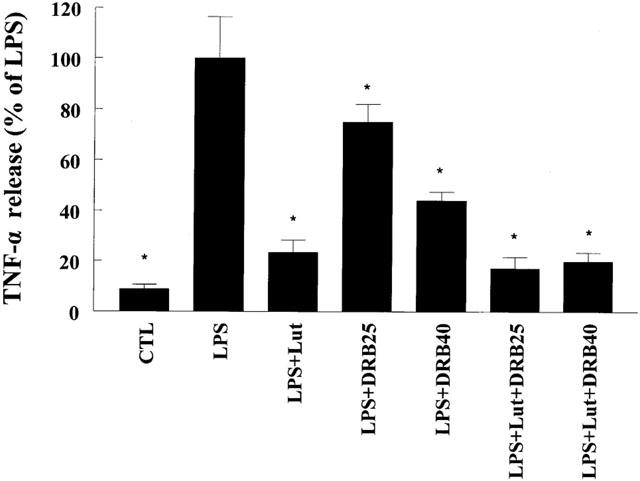
Inhibition of CK2 attenuates LPS-induced TNF-α release. Cells were pretreated for 60 min with vehicle or the indicated concentration of the pharmacological inhibitor of CK2 DRB (25 μM or 40 μM) in the presence and absence of luteolin (10 μM, 30 min), and then challenged with LPS (10 ng ml−1). After 24 h cells TNF-α was measured in the supernatants as described above. Data are presented as means±s.e.mean; n=10–16; *P<0.05 from LPS.
Discussion
The MAPK family of proteins comprises of three-tiered cascades involved in the regulation of multiple cellular processes ranging from gene expression to cell death (Chang & Karin, 2001; Davis, 2000). Exposure of mammalian cells to LPS has been shown to activate all known MAPK signalling cascades (Feng et al., 1999). In agreement to previous observations, exposure of RAW 264.7 cells to LPS caused a time-dependent phosphorylation of ERK1/2, p38 and JNK pathways. Interestingly, pretreament of cells with luteolin abolished the LPS-induced stimulation of ERK1/2 and p38, but not JNK, phosphorylation. The finding that pretreatment with luteolin also blocks phosphorylation of the MAPKKs MEK1/2 and MKK3/6 suggests that luteolin might act by inhibiting upstream activators of the ERK and p38 pathways. The potential upstream targets for luteolin include several tyrosine kinases, as it is known that tyrosine kinase inhibitors block LPS-induced MAPK activation (Novogrodsky et al., 1994) and we have demonstrated an inhibitory effect for luteolin on total tyrosine phosphorylation in response to LPS challenge (Xagorari et al., 2001).
The contribution of ERK1/2 and p38 pathways in stimulated TNF-α production in macrophages depends on the origin of macrophages, as well as the nature of the stimulus. For example, the MEK1/2 inhibitor PD98059 blocks LPS-induced TNF-α release from alveolar, but not non-pulmonary macrophages (Means et al., 2000). In addition, pharmacological inhibition of the p38 kinase in macrophage cell lines or targeted disruption of the MKK3 gene, that leads to reduced p38 activation, did not alter LPS-induced TNF-α production in peritoneal macrophages (Baldassare et al., 1999; Lu et al., 1999). In one report, treatment of freshly isolated peritoneal macrophages and 4-4 macrophages with the p38 inhibitor SB203580 resulted in increased TNF-α release (van-den-Blink et al., 2001). However, in other studies, blockade of the ERK1/2 or p38 pathways blocked LPS-stimulated TNF-α production (Kontoyiannis et al., 1999). To determine if inhibition of ERK1/2 and p38 phosphorylation by luteolin contributes to the reduction of TNF-α release in RAW 264.7 cells, we used specific inhibitors of the two pathways and determined their ability to block TNF-α release in our system. PD98059, a MEK1/2 inhibitor, when used at a concentration that blocks ERK1/2 phosphorylation, was ineffective in blocking LPS-induced TNF-α release. Similarly, SB203580 at 2.5 μM was also ineffective in reducing TNF-α production. It should be noted that although SB203580 has an IC50 for p38 in the nanomolar range, it is commonly used at 20–100 μM, in spite of reports demonstrating that at high concentrations SB203580 looses its specificity inhibiting PDK1 and enhancing NF-κB transcriptional activity by a non-specific effect on the ERK pathway (Lali et al., 2000). Concentrations of SB203580 up to 20 μM in our system caused a minimal inhibition (25%) in TNF-α release (data not shown). Interestingly, simultaneous inhibition of the two MAPK pathways, using a combination of SB203580 and PD98059, drastically reduced TNF-α release. Similar results have been obtained with alveolar macrophages, where activation of both ERK and p38 is necessary for the optimal TNF-α production (Carter et al., 1999). Since luteolin blocks LPS-induced phosphorylation of both MAPK pathways, its ability to inhibit TNF-α production could result from simultaneous inhibition of the ERK1/2 and p38 pathways.
LPS challenge of the RAW 264.7 macrophage cell line has been demonstrated to lead to activation of phosphatidylinositol 3-kinase (PI3-K) (Salh et al., 1998). While PI3-K has been mostly studied in the context of inhibition of apoptosis, there is evidence that PI3-K regulates cytokine gene expression in many cell types (Carpenter & Cantley, 1996). In addition, cross-talk between the MAPK and PI3-K pathways has been observed (Clauss et al., 2001; Madrid et al., 2001; Sutor et al., 1999). To determine whether inhibition of PI3-K contributes to the inhibitory action of luteolin on LPS-induced TNF-α release we used two pharmacological inhibitors of PI3-K. Cells pretreated with LY294002 prior to the LPS exposure showed reduced TNF-α release only at the highest concentration used. In addition, a chemically distinct PI3-K inhibitor, wortmannin, failed to block LPS-induced TNF-α release. Differences in the ability of the two PI3-K inhibitors, LY294002 and wortmannin, to block inducible molecule expression have been previously reported; exposure of RAW 264.7 to LY294002 abolished nitrite production in response to LPS, whereas wortmannin was ineffective (Salh et al., 1998). To help clarify the difference observed between the two PI3-K inhibitors, we used myricetin, a flavonoid known to inhibit PI3-K (Gamet-Payrastre et al., 1999). Pretreatment of cells with myricetin did not affect TNF-α production. The above observations taken together suggest that although luteolin is an inhibitor of PI3-K (Gamet-Payrastre et al., 1999), its action on LPS-induced TNF-α release is not related to its ability to block PI3-K.
We have previously shown that luteolin blocks Akt phosphorylation in response to LPS in RAW 264.7 macrophages (Xagorari et al., 2001). In mast cells, Akt activation stimulates transcriptional activity from the TNF-α promoter and production of TNF-α (Kitaura et al., 2000). Although Akt is a well accepted downstream target of PI3-K, activation of Akt in LPS-stimulated RAW 264.7 has been reported to be PI3-K-independent (Salh et al., 1998). To address the role of Akt in LPS-induced TNF-α release, cells were transiently transfected with a dominant negative form of Akt and exposed to LPS. Overexpression of K179M Akt did not alter LPS-induced TNF-α release, suggesting that inhibition of this kinase does not mediate the inhibitory action of luteolin on LPS-induced TNF-α production in this macrophage cell line.
While LY294002 is generally regarded as a specific PI3-K inhibitor, Davies et al. (2000) reported that it also inhibits CK2. Casein kinase 2 is a ubiquitously expressed ser/thr kinase with high basal activity, whose targets include enzymes involved in DNA, RNA and protein synthesis, transcription factors, signal transduction mediators, cytoskeletal and structural proteins (Guerra et al., 1999). Moreover, it has been suggested that CK2 promotes transition of inactive nucleosomes to the active conformation (Guo et al., 1998). In spite of the high basal activity, exposure of RAW 264.7 macrophages to LPS has been shown to lead to further activation of CK2 (Lodie et al., 1997). As LY294002 was the only PI3-K inhibitor that blocked TNF-α production, we postulated that its action might be due to CK2 inhibition rather than PI3-K inhibition. To determine the role of CK2 in LPS-induced TNF-α release we pretreated cells with the pharmacological inhibitor of CK2 DRB. Indeed, DRB-pretreated cells showed a concentration-dependent reduction in TNF-α production in response to LPS and co-treatment of cells with luteolin and DRB did not result in a greater inhibition than that observed with luteolin alone. This data, taken together with the observation that chrysin, a related flavonoid binds and inhibits CK2 (Critchfield et al., 1997), suggest that luteolin might reduce TNF-α production by inhibiting CK2. Direct evidence that luteolin is a pharmacological inhibitor of CK2 is offered by the observation that luteolin blocks more than 90% of CK2 activity in in vitro kinase reactions and abolishes CK2-induced p65 phosphorylation (T. Fotsis & E. Bagli, personal communication).
In conclusion, we have shown that luteolin interferes with multiple LPS-stimulated signaling cascades. Although luteolin blocks PI3-K activity and Akt phosphorylation (Gamet-Payrastre et al., 1999; Xagorari et al., 2001), inhibition of this pathway does not contribute to the inhibitory action of luteolin on LPS-induced TNF-α release. On the other hand, simultaneous blockade of ERK and p38, as well as CK2 inhibition by luteolin is linked to reduced production of TNF-α. The existence of multiple intracellular targets for luteolin can be explained by the fact that flavonoids compete with ATP binding and are thus able to inhibit several kinases (Middleton et al., 2001). Blockade of cascades involved in pro-inflammatory molecule expression by luteolin might be useful in preventing cytokine expression in pathophysiological conditions.
Acknowledgments
We wish to thank Dr W.C. Sessa for providing the dominant negative Akt. This study was supported by a grant from the Greek Secretariat of Research and Technology and by the Thorax Foundation.
Abbreviations
- CK2
casein kinase 2
- DRB
5,6-dichlororibifuranosylbenzimidasole
- ERK
extracellular signal-regulated kinase
- JNK
c-jun N-terminal kinase
- LPS
lipopolysaccharide
- Lut
luteolin
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- PI3-K
phosphatidylinositol 3-kinase
- TNF-α
tumour necrosis factor alpha
- WM
wortmannin
References
- AKIRA S., TAKEDA K., KAISHO T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- BALDASSARE J.J., BI Y., BELLONE C.J. The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J. Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- BAUMGARTNER J.D., CALANDRA T. Treatment of sepsis: past and future avenues. Drugs. 1999;57:127–132. doi: 10.2165/00003495-199957020-00001. [DOI] [PubMed] [Google Scholar]
- CARPENTER C., CANTLEY L. Phosphoinositide-3 kinase and the regulation of cell growth. Biochim Biophys Acta. 1996;1288:M11–M16. doi: 10.1016/0304-419x(96)00018-2. [DOI] [PubMed] [Google Scholar]
- CARTER A.B., MONICK M.M., HUNNINGHAKE G.W. Both Erk and p38 kinases are necessary for cytokine gene transcription. Am. J. Resp. Cell Mol. Biol. 1999;20:751–758. doi: 10.1165/ajrcmb.20.4.3420. [DOI] [PubMed] [Google Scholar]
- CHANG L., KARIN M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- CLAUSS M., SUNDERKOTTER C., SVEINBJORNSSON B., HIPPENSTIEL S., WILLUWEIT A., MARINO M., HAAS E., SELJELID R., SCHEURICH P., SUTTORP N., GRELL M., RISAU W. A permissive role for tumor necrosis factor in vascular endothelial growth factor-induced vascular permeability. Blood. 2001;97:1321–1329. doi: 10.1182/blood.v97.5.1321. [DOI] [PubMed] [Google Scholar]
- CRITCHFIELD J.W., COLIGAN J.E., FOLKS T.M., BUTERA S.T. Casein kinase II is a selective target of HIV-1 transcriptional inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6110–6115. doi: 10.1073/pnas.94.12.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES S.P., REDDY H., CAIVANO M., COHEN P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- DUMITRU C.D., CECI J.D., TSATSANIS C., KONTOYIANNIS D., STAMATAKIS K., LIN J.H., PATRIOTIS C., JENKINS N.A., COPELAND N.G., KOLLIAS G., TSICHLIS P.N. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–1083. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- FENG G.J., GOODRIDGE H.S., HARNETT M.M., WEI X.Q., NIKOLAEV A.V., HIGSON A.P., LIEW F.Y. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J. Immunol. 1999;163:6403–6412. [PubMed] [Google Scholar]
- GAMET-PAYRASTRE L., MANENTI S., GRATACAP M.P., TULLIEZ J., CHAP H., PAYRASTRE B. Flavonoids and the inhibition of PKC and PI 3-kinase. Gen. Pharmacol. 1999;32:279–286. doi: 10.1016/s0306-3623(98)00220-1. [DOI] [PubMed] [Google Scholar]
- GEPPERT T.D., WHITEHURST C.E., THOMPSON P., BEUTLER B. Lipopolysaccharide signals activation of tumor necrosis factor biosynthesis through the ras/raf-1/MEK/MAPK pathway. Mol. Med. 1994;1:93–103. [PMC free article] [PubMed] [Google Scholar]
- GERRITSEN M.E., CARLEY W.W., RANGES G.E., SHEN C.P., PHAN S.A., LIGON G.F., PERRY C.A. Flavonoids inhibit cytokine-induced endothelial cell adhesion protein gene expression. Am. J. Pathol. 1995;147:278–292. [PMC free article] [PubMed] [Google Scholar]
- GUERRA B., BOLDYREFF B., SARNO S., CESARO L., ISSINGER O.G., PINNA L.A. CK2: a protein kinase in need of control. Pharmacol. Ther. 1999;82:303–313. doi: 10.1016/s0163-7258(98)00064-3. [DOI] [PubMed] [Google Scholar]
- GUO C., DAVIES A.T., AHMED K. Dynamics of protein kinase CK2 association with nucleosomes in relation to transcriptional activity. J. Biol. Chem. 1998;273:13675–13680. doi: 10.1074/jbc.273.22.13675. [DOI] [PubMed] [Google Scholar]
- HOSHINO K., TAKEUCHI O., KAWAI T., SANJO H., OGAWA T., TAKEDA Y., TAKEDA K., AKIRA S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- KARIMA R., MATSUMOTO S., HIGASHI H., MATSUSHIMA K. The molecular pathogenesis of endotoxic shock and organ failure. Mol. Med. Today. 1999;5:123–132. doi: 10.1016/s1357-4310(98)01430-0. [DOI] [PubMed] [Google Scholar]
- KITAURA J., ASAI K., MAEDA-YAMAMOTO M., KAWAKAMI Y., KIKKAWA U., KAWAKAMI T. Akt-dependent cytokine production in mast cells. J. Exp. Med. 2000;192:729–740. doi: 10.1084/jem.192.5.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONTOYIANNIS D., PASPARAKIS M., PIZARRO T.T., COMINELLI F., KOLLIAS G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- KOTANIDOU A., XAGORARI A., BAGLI E., KITSANTA P., FOTSIS T., PAPAPETROPOULOS A., ROUSSOS C. Luteolin reduces LPS-induced lethal toxicity and expression of pro-inflammatory molecules in mice. Am. J. Resp. Crit. Care Med. 2002;165:818–823. doi: 10.1164/ajrccm.165.6.2101049. [DOI] [PubMed] [Google Scholar]
- LALI F.V., HUNT A.E., TURNER S.J., FOXWELL B.M. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 2000;275:7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- LODIE T.A., SAVEDRA R., GOLENBOCK D.T., VAN-BEVEREN C.P., MAKI R.A., FENTON M.J. Stimulation of macrophages by lipopolysaccharide alters the phosphorylation state, conformation, and function of PU.1 via activation of casein kinase II. J. Immunol. 1997;158:1848–1856. [PubMed] [Google Scholar]
- LU H.T., YANG D.D., WYSK M., GATTI E., MELLMAN I., DAVIS R.J., FLAVELL R.A. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999;18:1845–1857. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADRID L.V., MAYO M.W., REUTHER J.Y., BALDWIN A.S. Akt Stimulates the Transactivation Potential of the RelA/p65 Subunit of NF-kappa B through Utilization of the Ikappa B Kinase and Activation of the Mitogen-activated Protein Kinase p38. J. Biol. Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- MEANS T.K., PAVLOVICH R.P., ROCA D., VERMEULEN M.W., FENTON M.J. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J. Leuk. Biol. 2000;67:885–893. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- MIDDLETON E.J., KANDASWAMI C., THEOHARIDES T.C. The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease and Cancer. Pharmacol. Rev. 2001;52:673–751. [PubMed] [Google Scholar]
- NOVOGRODSKY A., VANICHKIN A., PATYA M., GAZIT A., OSHEROV N., LEVITZKI A. Prevention of lipopolysaccharide-induced lethal toxicity by tyrosine kinase inhibitors. Science. 1994;264:1319–1322. doi: 10.1126/science.8191285. [DOI] [PubMed] [Google Scholar]
- SALH B., WAGEY R., MAROTTA A., TAO J.S., PELECH S. Activation of phosphatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide production. J. Immunol. 1998;161:6947–6954. [PubMed] [Google Scholar]
- SUTOR S.L., VROMAN B.T., ARMSTRONG E.A., ABRAHAM R.T., KARNITZ L.M. A phosphatidylinositol 3-kinase-dependent pathway that differentially regulates c-Raf and A-Raf. J. Biol. Chem. 1999;274:7002–7010. doi: 10.1074/jbc.274.11.7002. [DOI] [PubMed] [Google Scholar]
- SWANTEK J.L., COBB M.H., GEPPERT T.D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol. Cell. Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN-DEN-BLINK B., JUFFERMANS N.P., TEN-HOVE T., SCHULTZ M.J., VAN-DEVENTER S.J., VAN-DER-POLL T., PEPPELENBOSCH M.P. p38 mitogen-activated protein kinase inhibition increases cytokine release by macrophages in vitro and during infection in vivo. J. Immunol. 2001;166:582–587. doi: 10.4049/jimmunol.166.1.582. [DOI] [PubMed] [Google Scholar]
- VASSALLI P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- WADSWORTH T.L., KOOP D.R. Effects of the wine polyphenolics quercetin and resveratrol on pro-inflammatory cytokine expression in RAW 264.7 macrophages. Biochem. Pharmacol. 1999;57:941–949. doi: 10.1016/s0006-2952(99)00002-7. [DOI] [PubMed] [Google Scholar]
- WRIGHT S.D., RAMOS R.A., TOBIAS P.S., ULEVITCH R.J., MATHISON J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- XAGORARI A., PAPAPETROPOULOS A., MAUROMATIS A., ECONOMOU M., FOTSIS T., ROUSSOS C. Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J. Pharmacol. Exp. Ther. 2001;296:181–187. [PubMed] [Google Scholar]