

Abstract
To identify the roles of endogenous kinins in prevention of myocardial infarction (MI), we performed the permanent ligation of coronary artery in rats.
The size of MI 12, 24, and 48 h after coronary ligation in kininogen-deficient Brown Norway Katholiek (BN-Ka) rats was significantly larger (49.7±0.2%, 49.6±2%, and 51.1±1%, respectively) than that of kinin-replete Brown Norway Kitasato (BN-Ki) rats (42±2%, 38.5±4%, and 41.5±1%).
Hoe140, a bradykinin (BK) B2 receptor antagonist injected (1.0 mg kg−1, i.v.) half an hour before, and every 8 h after, coronary ligation, significantly increased the size of MI in Sprague-Dawley rats. Aprotinin, a kallikrein inhibitor, which was infused intravenously (10,000 Units kg−1 h−1) with an osmotic mini-pump, significantly increased the size of an MI 24 h after ligation.
When evaluated using microspheres, the regional myocardial blood flow around the necrotic lesion in BN-Ka rats 6 h after ligation was reduced more than that in BN-Ki rats with MI by 41–46%. The same was true in Hoe140-treated BN-Ki rats.
FR190997, a nonpeptide B2 agonist, which was infused (10 μg kg−1 h−1) into the vena cava of BN-Ka rats for 24 h with an osmotic mini-pump, caused significant reduction in the size of MI (38±3%), in comparison with the size in vehicle solution-treated rats (51±3%). The size of MI in FR190997-treated BN-Ka rats was the same as in BN-Ki rats.
These results suggested that endogenous kinin has the capacity to reduce the size of MI via B2 receptor signalling because of the increase in regional myocardial blood flow around the ischaemic lesion.
Keywords: Myocardial infarction, bradykinin, FR190997, Hoe140, Brown Norway Katholiek rats
Introduction
It has been reported that angiotensin converting enzyme (ACE) inhibitors have many beneficial effects in cardiovascular disease (Linz et al., 1995). Occlusion of the coronary artery causes cardiomyocyte dysfunction and eventual tissue necrosis. ACE inhibitors are known to protect myocardium against ischaemia and to prevent ischaemia reperfusion injury (Fleetwood et al., 1991; Linz et al., 1986; van gilst et al., 1987). In particular, ACE inhibitors reduced the extent of cellular necrosis of the myocardium after coronary ligation (Ertl et al., 1982; Hock et al., 1985). ACE is the same enzyme as kininase II, one of the major kinin-degrading enzymes (Linz et al., 1995), so ACE inhibitors may accumulate bradykinin in the circulation or in the tissues where kinins were generated. Some beneficial effects of ACE inhibitors were reported to be cancelled by a kinin antagonist (Hartman et al., 1993).
The size of the myocardial infarctions (MI) of animals that had received a continuous infusion of kinin into the coronary artery before the coronary occlusion was reported to be reduced significantly (Martorana et al., 1990). Further, it was recently reported that delivery of the gene of tissue kallikrein, a kinin-generating enzyme, attenuated MI and reduced apoptosis, although delivery of kallikrein gene was performed 1 week before the induction of MI (Yoshida et al., 2000). Accordingly, it is probable that endogenous bradykinin may play an important role in the cardioprotective effect against MI, and that ACE inhibitors reduced MI via the accumulation of endogenous kinins. However, the mechanism of the protective effects of kinins against MI has not been fully elucidated.
Kinins are well known to dilate the resistance vessels and to increase the coronary blood flow (Linz et al., 1995). Thus, it is plausible that the endogenous kinins may inhibit the necrosis of the myocardium by increment of blood supply through this vasodilation. In the present experiment, we used kininogen-deficient mutant rats (Hayashi et al., 1993; Majima & Katori, 1995), which are incapable of generating kinin, to evaluate the contribution of endogenous kinins to the prevention of MI, and administered FR190997, a nonpeptide mimic of bradykinin, which is highly resistant to kinin-degrading enzymes and stimulates B2 receptors (Aramori et al., 1997; Majima et al., 2000). This study elucidates the mechanism of action of endogenous kinins in reducing the severity of MI by improving the regional blood flow in the myocardium.
Methods
Animals
Brown Norway Katholiek (BN-Ka) rats (Rattus norvegicus, BN/fMai) were initially obtained from the Katholieke Universiteit of Leuven, Belgium. Normal rats of the same strain, Brown Norway Kitasato (BN-Ki) rats were transferred from the Microbiological Association, Frederick, MD, U.S.A., and were kept at Kitasato University. All rats used were males, in most cases 10–12 weeks old, but in some experiments, Sprague-Dawley (SD) strain rats 6 weeks old (SLC, Hamamatsu, Japan) were used. The body weight of these animals was approximately 170–200 g. The size of these animals were adequate to produce the MI model (Sasaki et al., 1988; Kuribayashi et al., 1996).
The animals were housed at a constant humidity (60±5%) and temperature (25±1°C), and kept on a 12-h light/12-h dark cycle throughout the experiments. They were given normal rat chow (NMF, Oriental Yeast Corp., Tokyo, Japan). The number of animals (n) used for each experiment is stated in the corresponding section. The study was performed in accordance with the guidelines for animal experiments of Kitasato University School of Medicine.
Basal haemodynamics of BN-Ka rats was not different from those of normal BN-Ki rats, if they were fed with low sodium diet and tap water without high sodium (Majima & Katori, 1995). Furthermore, under such a condition, the weights of major organs in BN-Ka rats including hearts were not also different from those in BN-Ki rats.
Induction of myocardial infarction
MI was induced according to the method of our previous report (Kuribayashi et al., 1996). Briefly, rats were anaesthetized lightly with ether, and the thorax was opened. The left main coronary artery was ligated 1–2 mm below its origin with white silk suture thread (4-0, Natsume Ind., Tokyo). The heart was then replaced in the thorax, which was immediately closed.
Administration of drugs
A bradykinin antagonist, Hoe140 (1.0 mg kg−1, dissolved in physiological saline, Peptide Institute) (Wirth et al., 1991a) was injected intravenously under light ether anaesthesia half an hour before, and every 8 h after, coronary ligation. A B1 receptor antagonist, des Arg9-D-Arg[Hyp3, Thi5, D-Tic7, Oic8] bradykinin (des Arg10-[Hoe140]) (Wirth et al., 1991b) which was obtained from Peninsula Laboratories (San Carlos, CA, U.S.A.), was also administered (1.0 mg kg−1) in the same manner as Hoe140. This dose of Hoe140 was quite sufficient to block B2 receptor in vivo (Wirth et al., 1991a). In a separate experiment, a low dose of Hoe140 (0.3 mg kg−1) was also tested. A nonpeptide mimic of bradykinin, FR190997 (8-[2,6-dichloro-3-[N-[(E)-4-(N-methylcarbamoyl) cinnamidoacetyl]-N -methyl- amino]benzyloxy] -2 - methyl-4-(2-pyridylmethoxy)quinoline) (Aramori et al., 1997) (10 μg kg−1 h−1, suspended in 0.01 N HCl at a concentration of 2.5 mg ml; and itself a gift from Fujisawa Pharmaceutical Corp., Osaka, Japan), was administered immediately after coronary ligation for 24 h by continuous infusion into the vena cava (8 μl h−1) using a microosmotic pump (Alzet model 2001D, Alza Corp., Palo Alto, CA, U.S.A.), which was primed according to the protocol from Alza Corp., and was implanted into the subcutaneous tissue of the backs of the rats. The tip of the cannula to infuse FR190997 was placed in the vena cava of each animal. This compound was first dissolved in 0.1 N HCl, and then diluted 1 : 10 with physiological saline. Vehicle solution (saline containing HCl) was administered to control animals using the same types of pump and cannula, in order to evaluate the specific activity of FR190997. Aprotinin (10,000 unit kg−1 h−1, dissolved in 0.9% NaCl/H2O, Wako Pharmaceutical Corp., Osaka, Japan) (Majima et al., 1991) was administered from 30 min after coronary ligation for 24 h by continuous intravenous infusion to the cervical vein using a microosmotic pump (Alzet model 2001D) implanted subcutaneously in the back. Trypsin inhibitor from soybeans (SBTI) (0.4 mg kg−1 h−1, dissolved in 0.9% NaCl in H2O, Wako Pharmaceutical Corp., Osaka, Japan) (Majima et al., 1993) was also administered from 30 min after coronary ligation for 24 h by continuous intravenous infusion using a microosmotic pump (Alzet model 2001D).
Evaluation of infarction size
The infarction size of the hearts at 6, 12, 24 and 48 h after ligation were evaluated by the previously reported methods (Sasaki et al., 1988; Kuribayashi et al., 1996). The heart was rapidly excised and sliced into four transverse rings (rings A, B, C and D, starting from the base) at 2–2.5 mm intervals. Three transverse rings (rings B, C and D) of the hearts were incubated in a 1% triphenyltetrazolium chloride (TTC) solution in a 0.09 M phosphate buffer (pH 8.5; 37°C, 20 min). TTC is reduced to a red formazan by electron acceptance. This reaction does not occur in the areas in which the mitochondria or oxidative systems have been damaged or impaired. Thus, the lack of staining demarcated the injured areas from the intact areas. The stained and unstained areas of the basal surface of each ring were photographed with a digital camera, NV-DCF1 (Matsushita Electric Industrial Co., Ltd. Osaka, Japan), and their sizes were measured using NIH computer image software. The unstained areas were taken to be the infarct areas and were expressed as a percentage of the total cut area of the left ventricle (%LV).
Measurement of blood pressure
Mean arterial blood pressure (MBP) was determined for 1 h in conscious and unrestrained rats, as reported previously (Majima et al., 1993), 23 h after administration of the Hoe140 or FR190997. The rats were anaesthetized with light ether anaesthesia, and a polyethylene cannula (PE-10, Clay Adams, Parsippany, NJ, U.S.A.) was inserted into the abdominal aorta through the femoral artery 2 days before the experiments. The cannula was connected to a PE-50 cannula (Clay Adams, Parsippany, NJ, U.S.A.) and exteriorized in the interscapular region. A blood pressure transducer (TP-200T, Nihon Kohden, Tokyo, Japan) was attached to the other end of the intra-arterial cannula, and the mean arterial blood pressure was monitored on a polygraph (WS-641-G, Nihon Kohden, Tokyo, Japan) (Majima et al., 1993). Starting 30 min after the connection of the transducer, recordings were made for over 1 h in the rats, which were kept in separate cages.
Determination of a stable bradykinin metabolite,BK-(1-5), in the blood and in the myocardium
Six hours after the induction of MI, 5 ml of blood was collected from the carotid artery under light ether anaesthesia through a cannula into plastic tubes containing 20 ml of ice-cold absolute ethanol. After shaking, the mixture was centrifuged for 30 min at 3500×g at 0°C. The ethanol extracts (supernatants) were evaporated to dryness and were washed with diethylether three times to remove the lipids. The washed samples were dissolved in 4 ml of distilled water that had been acidified with 0.2 ml of 0.01 N HCl and were applied to a Sep-Pak C18 cartridge column (Waters Associates, Milford, MA, U.S.A.). After the samples had been washed with 12 ml of distilled water and 4 ml of 0.1 M acetic acid, BK and products of its degradation were eluted with 6 ml of 80% (v v−1) acetonitrile containing 0.1 M acetic acid (Kauker et al., 1984). The kinin fraction was dissolved in 800 μl of the assay buffer, and the levels of BK-(1-5) were determined with an enzyme-linked immunosorbent assay (ELISA) kit (Dainippon Pharmaceutical Co. Ltd., Osaka, Japan) that was developed by us (Majima et al., 1996). The levels of BK-(1-5) were measured in rats with MI that underwent sham thoracotomy operations only. To determine the heart tissue levels of BK and BK-(1-5), the heart was excised, and was put into the liquid nitrogen. Then, the frozen heart was pulverized within a stainless cylinder cooled with liquid nitrogen. The powder of the heart was immediately put into 5 ml of 80% ethanol without thawing and mixed well with homogenizer. After centrifugation at 1500×g for 30 min at 4°C, the supernatant was dried with reduced pressure. The residue was washed with ether and the levels of BK and BK-(1-5) were determined as mentioned above with ELISA.
Measurement of regional myocardial blood flow with colored microspheres
Six hours after the left main coronary artery was ligated, measurement of the regional myocardial blood flow was done with colored microspheres. Regional myocardial blood flow was determined with a technique (Kowallik et al., 1991) using 15 μm microspheres (Dye-trak, Triton Technologies Inc.). A polyethylene cannula (PE-10, Clay Adams, Parsippany, NJ, U.S.A.) was inserted into the carotid artery under light ether anesthesia and the other end of it was connected to a PE-50 cannula (Clay Adams, Parsippany, NJ, U.S.A.). A blood pressure transducer (TP-200T, Nihon Kohden, Tokyo, Japan) was attached to the other end of the latter cannula, and the arterial blood pressure waves were monitored on a polygraph (WS-641-G, Nihon Kohden, Tokyo, Japan) (Majima et al., 1993). During the monitoring of the pressure, the polyethylene cannula was inserted until a change from an arterial blood pressure wave to a left ventricle pressure wave occurred. For each measurement, approximately 3×105 microspheres suspended in 0.3 ml of saline (containing 0.02% Tween 80) were injected into the left ventricle through the polyethylene cannula. Microsphere injection was followed by flushing with 0.2 ml of saline. After microsphere injection, the rats were exsanguinated and their hearts were excised under ether anaesthesia. The atrium and right ventricle were removed. The heart was rapidly excised and sliced into four transverse rings (rings A, B, C and D, starting from the base) at 2–2.5 mm intervals. One transverse ring (ring B) of each heart was stained with TTC. TTC staining is reduced to a red formazan by electron acceptance. This reaction is lost in the areas in which mitochondria or oxidative systems have been damaged. Ring B was cut into quadrants, starting from the centre of the TTC-unstained area (indicated as clear area) as quadrant a and working clockwise (quadrants b, c and d) (Figure 5). One ml of a 4 M KOH solution containing 2% Tween 80 was added to each quadrant of ring B to digest the tissue. The plastic tubes were closed, and allowed to stand for 48 h at 25°C. The digest was removed in plastic tubes until only 50 μl of the sample remained. Five μl of that solution was placed in a counting chamber (improved Neubauer type), and the number of microspheres was counted under a microscope at a magnification of ×100.
Figure 5.
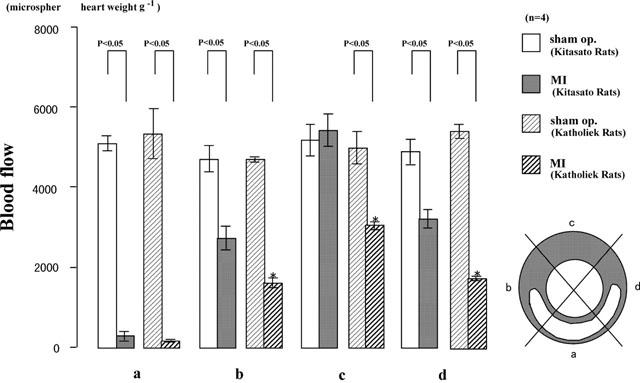
Measurement of regional myocardial blood flow with microspheres in kininogen-deficient Brown Norway Katholiek rats and normal Brown Norway Kitasato rats. Six hours after microsphere injection, the heart was excised and sliced into four transverse rings. One transverse ring (ring B, see the text) of the heart was stained by TTC. Ring B was cut into quadrants, starting from the center of the TTC-unstained area (indicated as clear area) as quadrant a and working clockwise (quadrants b, c and d). The number of microspheres in quadrants a, b, c, and d was counted under a microscope. Values are means±s.e.mean obtained from four rats. ANOVA was used to evaluate the significance of differences. *Compared with results from BN-Ki rats with coronary ligation.
Statistical analysis
Values were expressed as means±s.e.mean. Factorial ANOVA and repeated measure ANOVA with the post-hoc test were used to evaluate the significance of differences. For comparison between two groups, Student's t-test was used. A P value less than 0.05 was considered to be significant.
Results
Comparison of sizes of myocardial infarctions in kininogen-deficient Brown Norway Katholiek rats and normal Brown Norway Kitasato rats
The time course of MI size outlined by TTC (triphenyltetrazolium chloride) staining in normal BN-Ki rats is shown in Figure 1 (squares). Ligation of the coronary artery increased the unstained area, which reached a plateau at 12 h. The MI size 12, 24, and 48 h after coronary ligation in BN-Ki was 42±2%, 38.5±4%, and 41.5±1%, of left ventricle (LV) size, respectively (Figure 1, squares). The final size of MI in kininogen-deficient BN-Ka rats (Figure 1, triangles) was reached within 12 h, and was significantly (P<0.05) larger (49.7±0.2%, 49.6±2%, and 51.1±1% at 12, 24, and 48 h after ligation, respectively) than that observed in normal BN-Ki rats. One hour before the heart was excised at 24 h, MBP in BN-Ka rats was 99±3 mmHg (n=4), which was not statistically different from that determined in BN-Ki rats (101±3 mmHg, n=4). The mortality of BN-Ka rats (three rats dead among 12 rats) was not significantly different from that in BN-Ki rats (four rats dead among 13 rats).
Figure 1.
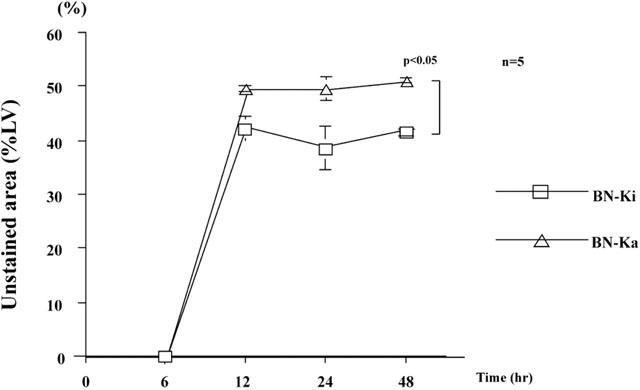
Time course of development of myocardial infarction in Brown Norway Katholiek rats and Brown Norway Kitasato rats. Myocardial infarction after coronary ligation was assessed using triphenyltetrazolium chloride (TTC) staining and expressed as a percentage of the unstained area of the left ventricle. Values are means±s.e.mean obtained from five rats. ANOVA was used to evaluate the significance of differences. LV, left ventricle; BN-Ka, Brown Norway Katholiek rats; BN-Ki, Brown Norway Kitasato rats; hr, hour(s).
Effect of Hoe140 on myocardial infarction size
Hoe140, a bradykinin (BK) B2 receptor antagonist, was tested in SD rats (Figure 2). When Hoe140 was injected (1 mg kg−1) half an hour before, and every 8 h after coronary ligation, it increased the infarct size to 44±3%, 45±2%, and 44±3% (n=5) at 12, 24, and 48 h after coronary ligation. This value was statistically significant (P<0.05) compared with that of vehicle-treated rats (Figure 2, right panel). However, des Arg10-[Hoe140], a bradykinin (BK) B1 receptor antagonist, did not affect the infarct size at 24 h (Figure 2, left panel).
Figure 2.
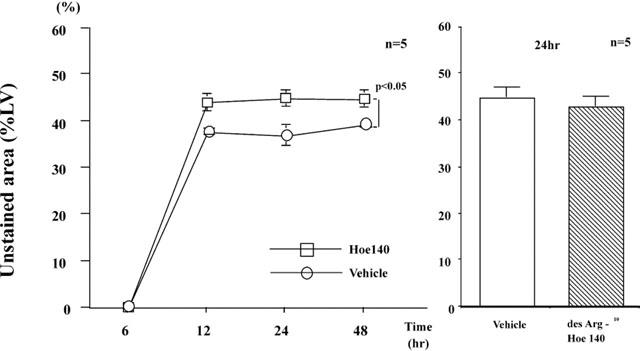
Effect of Hoe140, a B2 receptor antagonist, and des Arg10-[Hoe140], a B1 receptor antagonist, on the development of myocardial infarction in SD rats. Myocardial infarction after coronary ligation was assessed by triphenyltetrazolium chloride (TTC) staining and expressed as percentage of unstained area of the left ventricle. Values are means±s.e.mean obtained from five rats. ANOVA was used to evaluate the significance of differences. LV, left ventricle; Hoe140, SD rats receiving Hoe140; Vehicle, SD rats receiving vehicle; hr, hours.
When a low dose of Hoe140 was used, the infracted size 24 h after ligation was significantly increased to 43±2% (n=5), which was statistically larger than that in vehicle-treated rats.
One hour before the heart was excised at 24 h, MBP in Hoe140-treated rats was 102±2 mmHg (n=5), which was not statistically different from that determined in vehicle-treated rats.
Levels of BK-(1-5) in the circulation after myocardial infarction
The levels of BK-(1-5) in the blood collected from the carotid artery of SD rats with MI was determined by a specific ELISA 6 h after coronary ligation when the size of infarction was established (Figure 3). The levels of BK-(1-5) in rats with MI (183±32 pg ml−1) were increased significantly (P<0.05) compared with those in sham-operated rats (105±12 pg ml−1).
Figure 3.
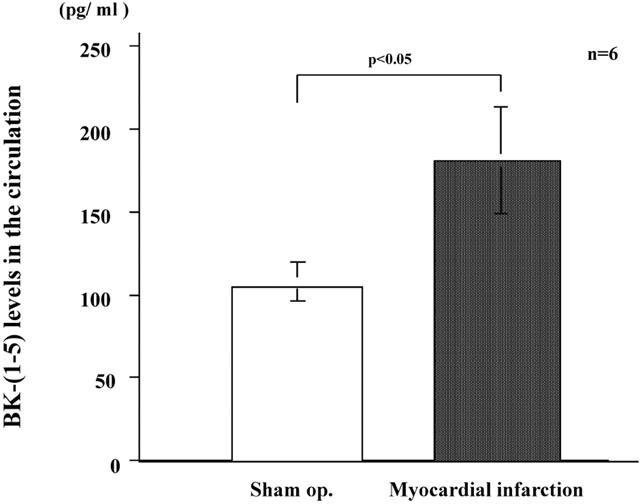
Kinin metabolite levels in the circulation. Blood was collected from the carotid artery 6 h after ligation, and BK-(1-5) levels were determined by a specific enzyme-linked immunosorbent assay. Values are means±s.e.mean obtained from six rats. The t-test was used to evaluate the significance of differences. Sham op., samples from sham-operated rats.
The tissue levels of both BK and BK-(1-5), determined in the excised hearts were less than the detection limit (90 pg heart−1) in sham-operated rats and rats with coronary ligation.
Effect of kallikrein inhibitors on myocardial infarction size
Aprotinin, a tissue and plasma kallikrein inhibitor, which was continuously infused with an osmotic mini-pump into the jugular vein of SD rats (50,000 Units kg−1 24 h−1) starting immediately after coronary ligation, significantly (P<0.05) increased the size of MI 24 h after ligation to 47±3%LV compared with that observed in vehicle-treated rats (Figure 4).
Figure 4.
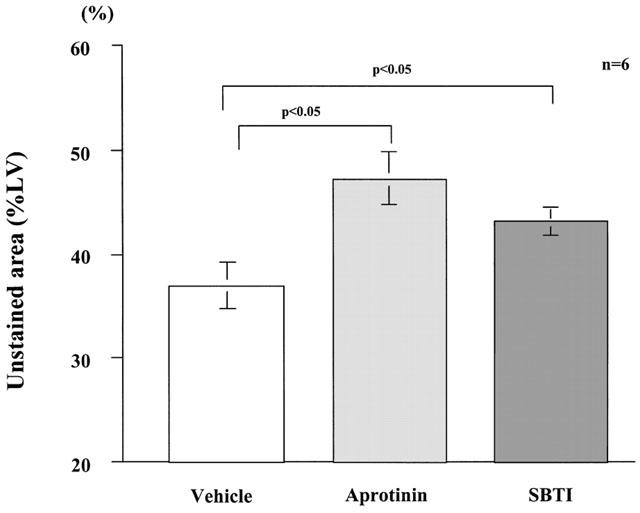
Effects of aprotinin and soybean trypsin inhibitor on the development of myocardial infarction in SD rats. Myocardial infarction after coronary ligation was assessed using triphenyltetrazolium chloride (TTC) staining and was expressed as percentage of unstained area of the left ventricle 24 h after coronary ligation. Values are means±s.e.mean obtained from six rats. ANOVA was used to evaluate the significance of differences. LV, left ventricle; aprotinin, SD rats receiving aprotinin; SBTI, SD rats receiving soybean trypsin inhibitor; Vehicle, SD rats receiving vehicle solution.
SBTI, a plasma kallikrein inhibitor, which was continuously infused with an osmotic mini-pump into a cervical vein (30,000 Units kg−1 24 h−1) starting immediately after coronary ligation, also significantly (P<0.05) increased the size of MI 24 h after ligation to 43±3%LV (Figure 4). However, the magnitude of the effect of SBTI was less than that of aprotinin.
Measurement of regional myocardial blood flow with microspheres in myocardial infarction of Brown Norway Katholiek rats and Brown Norway Kitasato rats
The numbers of microspheres in the left ventricle in quadrants a, b, c, and d were approximately 5000 pieces per g of heart weight in BN-Ki sham-operated rats (Figure 5). The numbers of microspheres in quadrants a, b, and d in BN-Ki rats 12 h after coronary occlusion were markedly reduced compared with those in sham-operated rats (Figure 5). There was no marked reduction in the numbers of microspheres in the opposite non-necrotic area (quadrant c) (Figure 5).
The numbers of microspheres in quadrants a, b, c, and d were reduced in BN-Ka rats with coronary ligation (Figure 5). The numbers of microspheres in the myocardium of BN-Ka rats with MI in quadrants b, c, and d, but not in quadrant a, were significantly reduced by 41%, 44%, and 46%, respectively, compared with those in BN-Ki rats with MI (Figure 5).
Measurement of regional myocardial blood flow with microspheres in myocardial infarction of Brown Norway Kitasato rats treated with Hoe140
Hoe140 (1.0 mg kg−1, dissolved in physiological saline), a bradykinin B2 receptor antagonist, injected half an hour before coronary ligation, significantly reduced the numbers of microspheres in sites b, c, and d (Figure 6), but no effect was seen on the number of microspheres in the myocardium in quadrant a, which bore a necrotic lesion (Figure 6).
Figure 6.
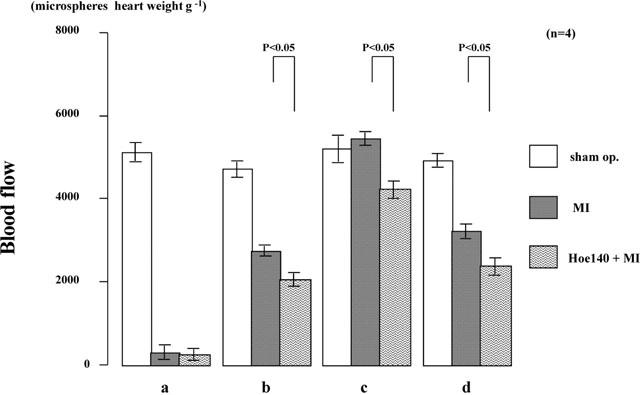
Effect of Hoe140 on regional myocardial blood flow in normal Brown Norway Kitasato rats. Regional blood flow was determined by the same method as that used in Figure 5. Values are means±s.e.mean obtained from four rats. ANOVA was used to evaluate the significance of differences. Sham op., rats that had sham operation; MI, rats with coronary ligation; Hoe140+MI, Hoe140-treated rats with coronary ligation.
Effect of FR 190997 on myocardial infarction size
FR190997, a nonpeptide mimic of bradykinin, which was infused (10 μg kg−1 h−1) continuously into the vena cava in BN-Ka rats for 24 h with an osmotic mini-pump, caused significant (P<0.05) reduction in the size of MI (38±3%LV), in comparison with the size in vehicle-treated rats (51±3%LV) (Figure 7). The size of MI in FR190997-treated BN-Ka rats was the same as that in normal BN-Ki rats (Figure 7). One hour before the heart was excised, the MBP in FR190997-treated BN-Ka rats was 89±1 mmHg (n=7), which was not significantly different from that in vehicle-treated BN-Ka rats.
Figure 7.
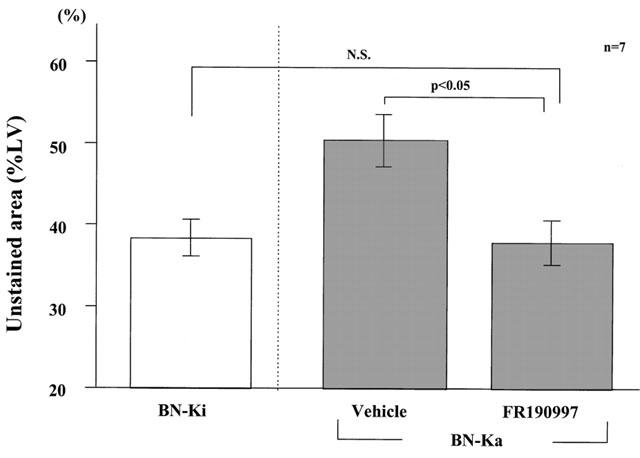
Effect of FR190997, a non-peptide B2 receptor agonist, on the development of a myocardial infarction in Brown Norway Katholiek rats. Myocardial infarction 24 h after coronary ligation was assessed by triphenyltetrazolium chloride (TTC) staining and expressed as percentage of unstained area of the left ventricle. Values are means with s.e.mean obtained from seven rats. ANOVA was used to evaluate the significance of differences. LV, left ventricle; BN-Ka, Brown Norway Katholiek rats; BN-Ki, Brown Norway Kitasato rats.
Discussion
Kinins are potent vasoactive proinflammatory peptides that are released from their precursor kininogens by kallikrein (Bhoola et al., 1992). The BN-Ka used in the present study exists only as an experimental animal, which is incapable of kinin generation. Plasma kininogens are secreted in the liver. The plasma levels of both high-molecular weight kininogen and low molecular weight kininogen were very low. This was attributable to the lack of the ability in this strain to secrete these kininogen moieties from the liver. One-point mutation of alanine163 to threonine in the kininogen moiety is responsible to this lack of secretion, although the hepatic cells of the mutant BN-Ka synthesized kininogens that do not differ in molecular weight (about 110 kDa) when assessed by electrophoresis (Hayashi et al., 1993). Comparison of BN-Ka with normal BN-Ki allowed us to study the pathophysiological roles of endogenous kinins in many situations (Majima & Katori, 1995). In the present study, we clarified the protective action of endogenous kinins in a model in which MI was induced by coronary occlusion, by making use of these mutant rats and their normal counterparts. The development of MI in BN-Ka was certainly enhanced throughout the experimental period, indicating that endogenous kinin has a protective action in this model. The size of the MI reached a plateau 12 h after coronary occlusion. Thus, the endogenous kinin generated from kininogens effectively prevented the development of MI for up to 12 h after the onset of myocardial ischaemia.
The endogenous kinin levels in the circulation 6 h after ligation, which was the developing stage of MI, was certainly elevated during the induction of MI, judging from the elevated levels in a stable metabolite of kinins, BK-(1-5). Kinin generation occurred independently in the two kinin systems in the body: the plasma kallikrein-high molecular weight kininogen system and the tissue kallikrein-low molecular weight kininogen system. It was believed that during ischaemia, plasma kallikrein was activated through the activation of factor XII (Pitt et al., 1969). The reduction in pH in the tissue by hypoxia was reported to activate this system. In fact, the elevation of kinin levels in the blood returning from the coronary circulation of the heart after an infarction was previously reported (Hashimoto et al., 1977). Previous investigation of the source of kinin has been poor. Tissue kallikrein together with plasma kallikrein were inhibited by aprotinin, although this inhibitor can inhibit other enzymes than kallikrein such as plasmin, whereas SBTI inhibited plasma kallikrein strongly. The effect of aprotinin was more pronounced in increasing the MI size than that of SBTI, and the possible involvement of tissue kallikrein in kinin formation was not ruled out completely in the present study. Kinins generated from the plasma kallikrein-high molecular weight kininogen system were reported to exert a protective effect on myocardial injury (Yang et al., 1997). However, another kinin generating system, the tissue kallikrein-low molecular weight kininogen system, may be responsible for these protective effects, since tissue kallikrein gene delivery attenuated MI together with apoptosis in rat coronary occlusion models (Yoshida et al., 2000). Expression of tissue kallikrein in the blood vessels and myocardium has frequently been reported (Nolly et al., 1993; 1994). Thus, the involvement of tissue kallikrein may not be ruled out in the present experiment. The significant increase in the tissue levels of BK and BK-(1-5) was not detected in the present experiment. These suggest that generating site of kinins may be restricted to the microcirculation rather than the interstitial tissues of myocardium.
To identify the BK receptors involved in this protective action of endogenous kinins, we administered the B2 receptor antagonist Hoe140 to rats whose left coronary artery was ligated. The results were essentially the same as those from mutant BN-Ka and those observed in aprotinin-treated rats, suggesting that B2 receptor signalling was a major pathway for the inhibition of MI in this model. The up-regulation of expression of B1 receptors was reported in the myocardium after infarction (Tschope et al., 2000). However, the lack of effect of des Arg10-[Hoe140], a B1 antagonist, suggest that the involvement of B1 receptor signalling may be negligible in the present experiment. It was reported that the cardioprotective action during coronary ischaemia was enhanced by ACE inhibitors, and this enhancement was abolished by bradykinin B2 receptor antagonists (Ito et al.,1997). Our present results also support the significance of B2 receptors without ACE inhibitor treatment.
How endogenous mediators or factors prevented ischaemic injury during coronary occlusion was extensively studied. Several potential factors such as adenosine (Liem et al., 2001; Miura et al., 2001), heart shock proteins (Hamilton et al., 2001), protein kinase C (Sharma & Singh, 2001; Matsumura et al., 2000), ATP-sensitive potassium channels (Toombs et al., 1993; Menasche et al., 1995), nitric oxide (Stefano et al., 2001), and prostaglandins (Shinmura et al., 2000) were believed to be involved in the preventive action against the development of MI. In addition to the above factors, it is believed that kinin may elicit this preventive action, because of its biological activities in increasing the coronary blood flow when infused directly into the coronary artery. To reveal the protective mechanisms of endogenous kinin, we determined the regional coronary blood flow using microsphores injected into the left ventricle (Kowallik et al., 1991). This approach enabled to determine the regional blood flow in small animals such as rats. The blood flow in the non-perfused regions was completely terminated after coronary occlusion in both BN-Ka and BN-Ki, since the blood supply from the left coronary artery was completely blocked. It is, however, quite interesting that, to judge from the significant reduction in the numbers of microspheres detected in BN-Ka and Hoe140-treated rats, the regional blood flow was maintained by endogenous kinin in the non-occluded regions such as those around the necrotic lesion and even in the quadrant opposite the infarction site, whose blood supply was maintained with the intact coronary artery. The endogenous kinins attenuate the MI, possibly by acting on the vasculature in the marginal zone of the area at risk, thus facilitating the blood flow from the non-ligated coronary artery or the marginal zone of the necrotic lesion.
Judging from the present results regarding the regional blood flow, the stimulation of B2 receptor signalling by coronary application will be expected to reduce the MI and subsequently save the patient from death by cardiac dysfunction. Supplementation of B2 agonist not only into the occluded coronary artery but also into the patient coronary artery will probably also reduce the size of MI. We previously reported that the first non-peptide B2 agonist, FR190997, dilated resistance vessels as did BK (Majima et al., 2000), but the vasodilating effect of FR190997 in vivo was considerably prolonged (Aramori et al., 1997; Majima et al., 2000). Furthermore, FR1909997 effectively prevented the development of hypertension in young spontaneously hypertensive rats (Majima et al., 2000). Therefore, it may be a powerful tool for the investigation of B2 receptor function and may be useful for therapy during MI. In fact, administration of FR190997 in BN-Ka rats caused significant reduction in the size of MI, in comparison with that in vehicle-treated rats. The size of MI in FR190997-treated BN-Ka rats was the same as that in BN-Ki rats. This suggested that B2 receptor signalling was the major signalling pathway for the attenuation of MI, and that a stable B2 receptor agonist, such as FR190997, is a good tool for the treatment of MI.
In conclusion, by using mutant rats and a selective B2 antagonist, we demonstrated that the endogenous kinins had a capacity for reducing the severity of MI through B2 receptor signalling. This protective action was attributable to the increase of regional blood flow around the ischaemic lesions. Pharmacological application of agents that can stimulate B2 receptor signaling may become useful tools for treating MI.
Acknowledgments
We express our thanks to Mr C.W.P. Reynolds for correcting the English of this manuscript, and to Ms M. Ogino for her technical assistance. This work was supported by research grants (#12470529 and #12670094), by “High-tech Research Center” grant, and by Academic Frontier Project grant from the Ministry of Education, Culture, Sports, Science and Technology, and was supported by an Integrative Research Program of the Graduate School of Medical Science, Kitasato University.
Abbreviations
- ACE
angiotensin converting enzyme
- BK
bradykinin
- BN-Ka
Brown Norway Katholiek
- BN-Ki
Brown Norway Kitasato
- LV
left ventricle
- MI
myocardial infarction
- MBP
mean blood pressure
- SD
Sprague-Dawley
- TTC
triphenyltetrazolium chloride
References
- ARAMORI I., ZENKOH J., MORIKAWA N., ASANO M., HATORI C., SAWAI H., KAYAKIRI H., SATOH S., INOUE T., ABE Y., SAWADA Y., MIZUTANI T., INAMURA N., NAKAHARA K., KOJO H., OKU T., NOTSU Y. Nonpeptide mimic of bradykinin with long-acting properties at the bradykinin B2 receptor. Mol. Pharmacol. 1997;52:16–20. doi: 10.1124/mol.52.1.16. [DOI] [PubMed] [Google Scholar]
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- ERTL G., KLONER R.A., ALEXANDER R.W., BRAUNWALD E. Limitation of experimental infarct size by an angiotensin-converting enzyme inhibitor. Circulation. 1982;65:40–48. doi: 10.1161/01.cir.65.1.40. [DOI] [PubMed] [Google Scholar]
- FLEETWOOD G., BOUTINET S., MEIER M., WOOD J.M. Involvement of the renin-angiotensin system in ischemic damage and reperfusion arrhythmias in the isolated perfused rat heart. J. Cardiovasc. Pharmacol. 1991;17:351–356. doi: 10.1097/00005344-199103000-00001. [DOI] [PubMed] [Google Scholar]
- HAMILTON K.L., POWERS S.K., SUGIURA T., KIM S., LENNON S., TUMER N., MEHTA J.L. Short-term exercise training can improve myocardial tolerance to I/R without elevation in heat shock proteins. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H1346–H1352. doi: 10.1152/ajpheart.2001.281.3.H1346. [DOI] [PubMed] [Google Scholar]
- HARTMAN J.C., WALL T.M., HULLINGER T.G., SHEBUSKI R.J. Reduction of myocardial infarct size in rabbits by ramiprilat: reversal by the bradykinin antagonist HOE 140. J. Cardiovasc. Pharmacol. 1993;21:996–1003. doi: 10.1097/00005344-199306000-00022. [DOI] [PubMed] [Google Scholar]
- HASHIMOTO K., HIROSE M., FURUKAWA S., HAYAKAWA H., KIMURA E. Changes in hemodynamics and bradykinin concentration in coronary sinus blood in experimental coronary artery occlusion. Jpn. Heart J. 1977;18:679–689. doi: 10.1536/ihj.18.679. [DOI] [PubMed] [Google Scholar]
- HAYASHI I., HOSHIKO S., MAKABE O., OH-ISHI S. A point mutation of alanine 163 to threonine is responsible for the defective secretion of high molecular weight kininogen by the liver of brown Norway Katholiek rats. J. Biol. Chem. 1993;268:17219–17224. [PubMed] [Google Scholar]
- HOCK C.E., RIBEIRO L.G., LEFER A.M.. Preservation of ischemic myocardium by a new converting enzyme inhibitor, enalaprilic acid, in acute myocardial infarction. Am. Heart J. 1985;109:222–228. doi: 10.1016/0002-8703(85)90587-3. [DOI] [PubMed] [Google Scholar]
- ITO K., ZHU Y.Z., ZHU Y.C., GOHLKE P., UNGER T. Contribution of bradykinin to the cardioprotective action of angiotensin converting enzyme inhibition in hypertension and after myocardial infarction. Jpn. J. Pharmacol. 1997;75:311–318. doi: 10.1254/jjp.75.311. [DOI] [PubMed] [Google Scholar]
- KAUKER M.L., CROFTON J.T., SHARE L., NASJLETTI A. Role of vasopressin in regulation of renal kinin excretion in Long-Evans and diabetes insipidus rats. J. Clin. Invest. 1984;73:824–831. doi: 10.1172/JCI111277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOWALLIK P., SCHULZ R., GUTH B.D. Measurement of reginal myocardial blood flow with multiple colored microspheres. Circulation. 1991;83:974–982. doi: 10.1161/01.cir.83.3.974. [DOI] [PubMed] [Google Scholar]
- KURIBAYASHI Y., KATORI M., MAJIMA M., YOSHIDA K. Inhibitory effects of a phosphate diester of alpha-tocopherol and ascorbic acid (EPC-K1) on myocardial infarction in rats. Int. J. Tissue React. 1996;18:73–79. [PubMed] [Google Scholar]
- LIEM D.A., VAN DEN DOEL M.A., DE ZEEUW S., VERDOUW P.D., DUNCKER D.J. Role of adenosine in ischemic preconditioning in rats depends critically on the duration of the stimulus and involves both A(1) and A(3) receptors. Cardiovasc. Res. 2001;51:701–708. doi: 10.1016/s0008-6363(01)00321-2. [DOI] [PubMed] [Google Scholar]
- LINZ W., SCHOLKENS B.A., HAN Y.F. Beneficial effects of the converting enzyme inhibitor, ramipril, in ischemic rat hearts. J. Cardiovasc. Pharmacol. 1986;8:S91–S99. doi: 10.1097/00005344-198600101-00017. [DOI] [PubMed] [Google Scholar]
- LINZ W., WIEMER G., GOHLKE P., UNGER T., SCHOLKENS B.A. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol. Rev. 1995;47:25–49. [PubMed] [Google Scholar]
- MAJIMA M., HAYASHI I., INAMURA N., FUJITA T., OGINO M. A nonpeptide mimic of bradykinin blunts the development of hypertension in young spontaneously hypertensive rats. Hypertension. 2000;35:437–442. doi: 10.1161/01.hyp.35.1.437. [DOI] [PubMed] [Google Scholar]
- MAJIMA M., KATORI M. Approaches to the development of novel antihypertensive drugs: crucial role of the renal kallikrein-kinin system. Trends Pharmacol. Sci. 1995;16:239–246. doi: 10.1016/s0165-6147(00)89033-1. [DOI] [PubMed] [Google Scholar]
- MAJIMA M., KATORI M., HANAZUKA M., MIZOGAMI S., NAKANO T., NAKAO Y., MIKAMI R., URYU H., OKAMURA R., MOHSIN S.S. Suppression of rat deoxycorticosterone-salt hypertension by kallikrein-kinin system. Hypertension. 1991;17:806–813. doi: 10.1161/01.hyp.17.6.806. [DOI] [PubMed] [Google Scholar]
- MAJIMA M., NISHIYAMA K., IGUCHI Y., YAO K., OGINO M., OHNO T., SUNAHARA N., KATOH K., TATEMICHI N., TAKEI Y., KATORI M. Determination of bradykinin-(1-5) in inflammatory exudate by a new ELISA as a reliable indicator of bradykinin generation. Inflamm. Res. 1996;45:416–423. doi: 10.1007/BF02252938. [DOI] [PubMed] [Google Scholar]
- MAJIMA M., YOSHIDA O., MIHARA H., MUTO T., MIZOGAMI S., KURIBAYASHI Y., KATORI M., OH-ISHI S. High sensitivity to salt in kininogen-deficient brown Norway Katholiek rats. Hypertension. 1993;22:705–714. doi: 10.1161/01.hyp.22.5.705. [DOI] [PubMed] [Google Scholar]
- MARTORANA P.A., KETTENBACH B., BREIPOHL G., LINZ W., SCHOLKENS B. Reduction of infarct size by local angiotensin-converting enzyme inhibition is abolished by a bradykinin antagonist. Eur. J. Pharmacol. 1990;182:395–396. doi: 10.1016/0014-2999(90)90301-l. [DOI] [PubMed] [Google Scholar]
- MATSUMURA K., KOMORI S., TAKUSAGAWA M., OSADA M., TANABE F., ITOH M., TAMURA K. Protein kinase C is involved in cardioprotective effects of ischemic preconditioning on infarct size and ventricular arrhythmia in rats in vivo. Mol. Cell Biochem. 2000;214:39–45. doi: 10.1023/a:1007119622322. [DOI] [PubMed] [Google Scholar]
- MENASCHE P., KEVELAITIS E., MOUAS C., GROUSSET C., PIWNICA A., BLOCH G. Preconditioning with potassium channel openers. A new concept for enhancing cardioplegic protection. J. Thorac. Cardiovasc. Surg. 1995;110:1606–1613. doi: 10.1016/S0022-5223(95)70020-X. [DOI] [PubMed] [Google Scholar]
- MIURA T., KAWAMURA S., TATSUNO H., IKEDA Y., MIKAMI S., IWAMOTO H., OKAMURA T., IWATATE M., KIMURA M., DAIRAKU Y., MAEKAWA T., MATSUZAKI M. Ischemic preconditioning attenuates cardiac sympathetic nerve injury via ATP-sensitive potassium channels during myocardial ischemia. Circulation. 2001;104:1053–1058. doi: 10.1161/hc3501.093800. [DOI] [PubMed] [Google Scholar]
- NOLLY H., CARBINI L.A., SCICLI G., CARRETERO O.A., SCICLI A.G. A local kallikrein-kinin system is present in rat hearts. Hypertension. 1994;23:919–923. doi: 10.1161/01.hyp.23.6.919. [DOI] [PubMed] [Google Scholar]
- NOLLY H., CARRETERO O.A., SCICLI A.G. Kallikrein release by vascular tissue. Am. J. Physiol,. 1993;265:H1209–H1214. doi: 10.1152/ajpheart.1993.265.4.H1209. [DOI] [PubMed] [Google Scholar]
- PITT B., MASON J., CONTI C.R., COLMAN R.W. Observations on the plasma kallikrein system during myocardial ischemia. Trans. Assoc. Am. Physicians. 1969;82:98–108. [PubMed] [Google Scholar]
- SASAKI K., UENO A., KATORI M., KIKAWADA R. Detection of leukotriene B4 in cardiac tissue and its role in infarct extension through leucocyte migration. Cardiovasc. Res. 1988;22:142–148. doi: 10.1093/cvr/22.2.142. [DOI] [PubMed] [Google Scholar]
- SHARMA A., SINGH M. Protein kinase C activation and cardioprotective effect of preconditioning with oxidative stress in isolated rat heart. Mol. Cell Biochem. 2001;219:1–6. doi: 10.1023/a:1011038531656. [DOI] [PubMed] [Google Scholar]
- SHINMURA K., TANG X.L., WANG Y., XUAN Y.T., LIU S.Q., TAKANO H., BHATNAGAR A., BOLLI R. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEFANO G.B., NEENAN K., CADET P., MAGAZINE H., BILFINGER T.V. Ischemic preconditioning – an opiate constitutive nitric oxide molecular hypothesis. Med. Sci. Monit. 2001;7:1357–1375. [PubMed] [Google Scholar]
- TOOMBS C.F., MOORE T.L., SHEBUSKI R.J. Limitation of infarct size in the rabbit by ischaemic preconditioning is reversible with glibenclamide. Cardiovasc. Res. 1993;27:617–622. doi: 10.1093/cvr/27.4.617. [DOI] [PubMed] [Google Scholar]
- TSCHOPE C., HERINGER-WALTHER S., KOCH M., SPILLMANN F., WENDORF M., LEITNER E., SCHULTHEISS H.P., WALTHER T. Upregulation of bradykinin B1-receptor expression after myocardial infarction. Br. J. Pharmacol. 2000;129:1537–1538. doi: 10.1038/sj.bjp.0703239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN GILST, W.H., VAN WIJNGAARDEN J., SCHOLTENS E., DE GRAEFF P.A., DE LANGEN C.D., WESSELING H. Captopril-induced increase in coronary flow: an SH-dependent effect on arachidonic acid metabolism. J. Cardiovasc. Pharmacol. 1987;9:S31–S36. [PubMed] [Google Scholar]
- WIRTH K., BREIPOHL G., STECHL J., KNOLLE J., HENKE S., SCHOLKENS B. DesArg9-D-Arg[Hyp3,Thi5,D-Tic7,Oic8]bradykinin (desArg10-[Hoe140]) is a potent bradykinin B1 receptor antagonist. Eur. J. Pharmacol. 1991b;205:217–218. doi: 10.1016/0014-2999(91)90824-a. [DOI] [PubMed] [Google Scholar]
- WIRTH K., HOCK F.J., ALBUS U., LINZ W., ALPERMANN H.G., ANAGNOSTOPOULOS H., HENK S., BREIPOHL G., KONIG W., KNOLLE J. Hoe 140 a new potent and long acting bradykinin-antagonist: in vivo studies. Br. J. Pharmacol. 1991a;102:774–777. doi: 10.1111/j.1476-5381.1991.tb12249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X.P., LIU Y.H., SCICLI G.M., WEBB C.R., CARRETERO O.A. Role of kinins in the cardioprotective effect of preconditioning: study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen-deficient rats. Hypertension. 1997;30:735–740. doi: 10.1161/01.hyp.30.3.735. [DOI] [PubMed] [Google Scholar]
- YOSHIDA H., ZHANG J.J., CHAO L., CHAO J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Hypertension. 2000;35:25–31. doi: 10.1161/01.hyp.35.1.25. [DOI] [PubMed] [Google Scholar]