

Abstract
The present study addressed whether endothelium-dependent vasodilatation evoked by acetylcholine and flow are mediated by the same mechanisms in isolated rat mesenteric small arteries, suspended in a pressure myograph for the measurement of internal diameter.
In pressurized arterial segments contracted with U46619 in the presence of indomethacin, shear stress generated by the flow evoked relaxation. Thus, in endothelium-intact segments low (5.1±0.6 dyn cm−2) and high (19±2 dyn cm−2) shear stress evoked vasodilatations that were reduced by, respectively, 68±11 and 68±8% (P<0.05, n=7) by endothelial cell removal. Acetylcholine (0.01–1 μM) evoked concentration-dependent vasodilatation that was abolished by endothelial cell removal.
Incubation with indomethacin alone did not change acetylcholine and shear stress-evoked vasodilatation, while the combination of indomethacin with the nitric oxide (NO) synthase inhibitor, NG,NG-asymmetric dimethyl-L-arginine (ADMA 1 mM), reduced low and high shear stress-evoked vasodilatation with, respectively, 52±15 and 58±10% (P<0.05, n=9), but it did not change acetylcholine-evoked vasodilatation.
Inhibition of Ca2+-activated K+ channels with a combination of apamin (0.5 μM) and charybdotoxin (ChTX) (0.1 μM) did not change shear stress- and acetylcholine-evoked vasodilatation. In the presence of indomethacin and ADMA, the combination of apamin (0.5 μM) and ChTx (0.1 μM) increased contraction induced by U46619, but these blockers did not change the vasodilatation evoked by shear stress. In contrast, acetylcholine-evoked vasodilatation was abolished by the combination of apamin and charybdotoxin.
In the presence of indomethacin, the tyrosine kinase inhibitor, herbimycin A (1 μM), inhibited low and high shear stress-evoked vasodilatation with, respectively, 32±12 and 68±14% (P<0.05, n=8), but it did not change vasodilatation induced by acetylcholine. In the presence of indomethacin and ADMA, herbimycin A neither changed shear stress nor acetylcholine-evoked vasodilatation.
The present study suggests that Ca2+-activated K+ channels sensitive for the combination of apamin and ChTx are involved in acetylcholine-evoked, mainly non-NO nonprostanoid factor-mediated, vasodilatation, while an Src tyrosine kinase plays a role for flow-evoked NO-mediated vasodilatation in rat mesenteric small arteries.
Keywords: Flow, acetylcholine, endothelium, resistance arteries, rat, herbimycin A
Introduction
The vascular endothelium plays an important role in the regulation of vascular tone through release of nitric oxide (NO) and prostaglandins. In addition, there are endothelium-dependent vasodilatation resistant to the inhibition of cyclooxygenase and NO synthase (NOS), non-NO, nonprostanoid endothelium-derived hyperpolarizing factor (EDHF)-type relaxation, which is most pronounced in the distal part of the systemic arterial circulation (Parsons et al., 1994; Hwa et al., 1994; Shimokawa et al., 1996; Buus et al., 2000; McGuire et al., 2001; Busse et al., 2002). Several candidates for the EDHF-type relaxation have been suggested to mediate agonist-induced relaxation of small arteries such as potassium ions (Edwards et al., 1998), hydrogen peroxide (H2O2) (Matoba et al., 2000), and products of the cytochrome P450 pathway (Fulton et al., 1992; Bauersachs et al., 1994; Fisslthaler et al., 1999; Bolz et al., 2000). Other studies have attributed agonist-evoked non-NO nonprostanoid relaxation to myoendothelial gap junction communication (Chaytor et al., 1998; Yamamoto et al., 1999;Sandow & Hill, 2000; Coleman et al., 2001; Taylor et al., 2001). Elevation of shear stress evokes NO-mediated relaxation in large systemic arteries (Rubanyi et al., 1986; Cooke et al., 1991), coronary small arteries (Muller et al., 1999) and human subcutaneous arteries (Paniagua et al., 2001), but it was shown to lead to EDHF-type vasodilatation in rat mesenteric arteries (Matrougui et al., 1997; Takamura et al., 1999; Izzard & Heagerty, 1999) and human coronary arterioles (Miura et al., 2001). In human coronary arterioles, shear stress-evoked vasodilatation is attributed to cytochrome P450 metabolites (Miura et al., 2001), but the non-NO nonprostanoid EDHF-type vasodilatation is not fully characterized in mesenteric small arteries and it has not been addressed whether acetylcholine and shear stress is causing vasodilatation by the same endothelium-dependent mechanism in small arteries.
The inhibition of small- and intermediate-conductance Ca2+-activated K+ channels by the combination of apamin and charybdotoxin (ChTx) has been considered as a unique characteristic of the non-NO nonprostanoid-type relaxations (Edwards et al., 1998; Buus et al., 2000), and hyperpolarization evoked by agonists in small arteries (Zygmunt & Högestatt, 1996; Edwards et al., 1998; Yamamoto et al., 1999). A general blocker of Ca2+-activated K+ channels, tetraethylammonium, and ChTx was also found to inhibit the non-NO nonprostanoid vasodilatation and hyperpolarization induced by flow in coronary arteries (Dube & Canty, 2001; Miura et al., 2001), while the combination of apamin and ChTx only caused partial inhibition of flow-induced vasodilatation in rat mesenteric arteries (Takamura et al., 1999). Therefore, these studies could suggest that flow activates another relaxation mechanism than acetylcholine. Agonist activation of isolated endothelial cells by acetylcholine is dependent on rises in intracellular Ca2+ (Nilius & Droogmans, 2001), while shear stress-evoked endothelial cell activation is partly dependent of Ca2+ and on the activation of tyrosine kinase or serine/threonine kinase (Corson et al., 1996; Fleming et al., 1998; Dimmeler et al., 1999). However, in small arteries it has not been addressed whether different kinds of endothelial cell activation such as by the endothelium-dependent agonist, acetylcholine, and flow lead to vasodilatation mediated by distinct endothelium-derived factors.
The aim of the present study was to clarify whether acetylcholine- and flow-induced vasodilatation is mediated by the same mechanisms in rat mesenteric small arteries. For this purpose, the effect of blockers of Ca2+-activated K+ channels and Src tyrosine kinase as well as inhibitors of cyclooxygenase and NO synthase were evaluated on flow- and acetylcholine-evoked vasodilatation in pressurized vascular segments.
Methods
The 12- to 16-week-old male Wistar rats were killed by a blow to the head followed by exsanguination. The procedures were in accordance with Danish Animal Law and regulations. The mesenteric vascular bed was removed and throughout the subsequent dissection, the tissue was bathed in cold physiological salt solution (PSS, 4°C) of the following composition (mM): CaCl2 1.6, NaCl 119, KCl 4.7, glucose 5.5, MgSO4·7H2O 1.17, NaHCO3 25, KH2PO4 1.18, and ethylenediaminetetraacetic acid (EDTA) 0.026. The solution was gassed with 5% CO2 in air to maintain pH at 7.4.
Flow-induced vasodilatation
Third-order mesenteric arteries were isolated by microdissection. After dissection arterial segments with a length of 1.5–3 mm were transferred to the chamber of a pressure myograph for cannulation (Danish Myotechnology, Aarhus, Denmark (Buus et al., 1994). At this stage, the chamber was filled with PSS equilibrated with room air at ambient temperature, and the perfusion line was filled with PSS. The chamber contained two glass pipettes (tip outer diameter ∼120–130 μm): one was fixed and the other was mounted on a manipulator allowing adjustment of the vessel length. The vessel was cannulated with the inflow pipette and tied by a surgical thread. The inflow pressure was elevated to 15–20 mmHg to wash out blood, and the other end of the vessel was cannulated with the outflow pipette. After cannulation, the pressure myograph was transferred to the stage of an inverted microscope, the pressure was elevated to 30 mmHg, and the myograph bath was heated to 37°C. Then the pressure was elevated to 80 mmHg, and the axial length was adjusted to eliminate any buckling of the preparation. When pressurized, the vessel segment was longitudinally stretched to 110% of the initial passive length at 80 mmHg. Under an inverted microscope (Telaval 31, Zeiss) the preparation was examined for leaks, which were easily identified by outflow of solution.
The middle of the vessel segment was viewed through the inverted microscope (magnification × 100), and its inner and outer diameter was measured by a videomicroscope technique. The signal from a video camera attached to the inverted microscope was fed to a frame grabber and then to a dimension-analyzing program (VesselView, Danish Myotechnology, Aarhus, Denmark) allowing continuous sampling of diameters and pressures at 3 Hz. Hydrostatic pressures of both inlet and outlet reservoirs were measured by pressure transducers connected to the perfusion line on the inlet and outlet side, respectively.
The arterial segment was exposed to U46619 (0.1 μM) added both intra- and extraluminally, since we observed that intraluminal washout contributed to the vasodilatation obtained by increasing flow. When the contraction was stable, acetylcholine (1 μM) was added extraluminally. The following criteria were applied to accept an arterial segment for the investigation of endothelium-dependent vasodilatation: (1) The contractile response induced by U46619 should reduce vessel diameter with at least 25%. (2) Relaxation evoked by acetylcholine (1 μM) above 50%. (3) If an air bubble passed the arterial segment, it led to exclusion of the experiment.
To investigate the role of the endothelial cell layer, a first response to flow and acetylcholine was obtained in the U46619-contracted artery. Flow was generated by suction with a peristaltic pump and flow was adjusted in the ranges of either 25–50 μl or 100–200 μl min−1, which correspond to shear stress levels of, respectively, 4 and 16 dyn cm−2 in the mesenteric small arteries. Shear stress was calculated from the following equation:
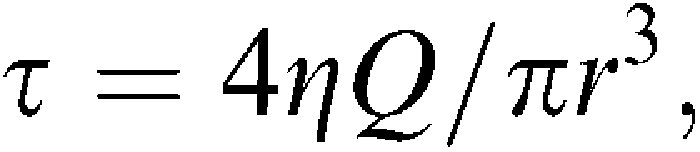 |
where r is the internal vessel diameter, Q the average flow velocity, and η the viscosity. In between the two responses to flow, the pump was stopped allowing the preconstriction to regain. The latter procedure was included, since we observed that very high flow, corresponding to 30–50 dyn cm−2, generated irreversible damage to the vascular wall. The suction from the peristaltic pump introduced a small pressure variation with an amplitude of 3 mmHg and frequency of 0.04–0.3 Hz. The segment was bubbled with small air bubbles, allowed to dilate and was constricted with U46619 (0.1 μM) and acetylcholine (1 μM) was added. Lack of vasodilatation to acetylcholine was taken as evidence of successful removal of the endothelial cell layer. A second response to flow and acetylcholine was obtained, and finally the segment was dilated with papaverine (100 μM).
For characterization of the mechanisms involved in the endothelium-dependent vasodilatation by flow and acetylcholine, the arterial segment was constricted with U46619 applied both extra- and intraluminally. When the constriction induced by U46619 was stable a first response was obtained for flow generating a shear stress corresponding to 4 and 16 dyn cm−2, and then acetylcholine (0.01–1 μM) was added. The preparation was washed and incubated with either indomethacin (3 μM), ADMA (1 mM), indomethacin and ADMA, or indomethacin and herbimycin (10 μM) for 20 min, and a second response was obtained for flow and acetylcholine. In another series of experiments, indomethacin and ADMA were present throughout the experiments, and a first response for flow and acetylcholine was obtained, after which a second response was obtained either in the absence of drugs or after incubation with apamin and ChTx, barium chloride (BaCl2,100 μM), or herbimycin A.
Drugs
The following drugs were used: acetylcholine HCl, indomethacin, asymmetric dimethyl L-arginine (ADMA), herbimycin A, 9,11-di-deoxy-11α, 9α-epoxymethano prostaglandin F2α (U46619), noradrenaline HCl, and phenylarsine oxide were from Sigma, U.S.A. Apamin and ChTx were from Latoxan, France, and BaCl2 from Merck, Germany.
Noradrenaline was prepared in 0.25 N HCl and further diluted in distilled water. U46619 was dissolved in 50% ethanol and further diluted in distilled water. The other drugs were dissolved in distilled water. None of the solvents, in the concentration applied, had any effect on the preparations.
Analysis of data
All responses are expressed as mean±s.e.m., where n equals the number of rats. Relaxations are expressed as changes in internal diameter or area under the relaxation response was calculated and expressed as changes in internal diameter (μm) with time (s) (GraphPad Prism, San Diego, CA, U.S.A.). The differences between means were analyzed with two-way ANOVA and complementary Bonferroni t-test. A value of P<0.05 was considered to indicate statistical significance.
Results
Flow-evoked vasodilatation
A total of 60 vessels were mounted, 43 segments were accepted, and 17 arteries excluded because of lack of response to flow (n=9), destruction by an air bubble passing the artery either accidently (n=5) or in connection with attempts to remove the endothelial cell layer (n=3). The average diameter of the mounted and accepted arteries was 353±10 μM, and U46619 constricted the arteries to 198±10 μM (n=43). In pressurized arterial segments contracted with U46619 in the presence of indomethacin, shear stress generated by the flow evoked relaxation. Thus, in endothelium-intact segments low (5.1±0.6 dyn cm−2) and high (19±2 dyn cm−2) shear stress evoked vasodilatations that were reduced by, respectively, 68±11 and 68±8% (P<0.05, n=7) by endothelial cell removal (Figure 1 and Figure 2a). Acetylcholine (0.01–1 μM) evoked concentration-dependent vasodilatation that was abolished by endothelial cell removal (Figure 1b and Figure 2b). Incubation with indomethacin (3 μM) did not change vasodilatations induced by flow or by acetylcholine (Figure 3a,b). In the presence of indomethacin, incubation with an inhibitor of NO synthase, ADMA (1 mM), reduced low and high shear stress-evoked vasodilatation with, respectively, 52±15 and 58±10% (P<0.05, n=9; Figure 4a). However, acetylcholine-evoked vasodilatation was largely unchanged in the presence of ADMA, although Student's t-test revealed significant differences comparing 10−7 M acetylcholine relaxation obtained in the absence and presence of ADMA (Figure 4b).
Figure 1.

Flow and acetylcholine-induced vasodilatation in rat mesenteric artery. Original traces showing the diameter changes in (a) endothelium-intact and (b) endothelium-denuded rat mesenteric artery contracted with U46619 (0.1 μM), and relaxed with flow corresponding to 4 and 16 dyn cm−2, and then with increasing concentrations of acetylcholine (ACh). The experiments were performed in the presence of indomethacin (3 μM).
Figure 2.

Endothelial cell removal abolished acetylcholine vasodilatation and inhibited flow-induced vasodilatation. Area under the vasodilatation curve (AUC) calculated for each response to (a) flow and (b) acetylcholine in endothelium-intact and -denuded arterial preparations. Results are mean±s.e.m. of seven preparations. The experiments were performed in the presence of indomethacin (3 μM). Differences in responses evaluated by two-way analysis of variance: *P<0.05 versus endothelium-intact preparations; vasodilatation responses were dependent on the magnitude of shear stress and acetylcholine concentration applied.
Figure 3.

Flow- and acetylcholine-induced vasodilatation resistant to the inhibition of cyclooxygenase. Area under the vasodilatation curve (AUC) calculated for each response to (a) flow and (b) acetylcholine obtained in the absence and the presence of indomethacin (3 μM). Results are mean±s.e.m. of five preparations.
Figure 4.

NG, NG-asymmetric dimethyl-L-arginine (ADMA) inhibits flow-evoked vasodilatation in rat mesenteric arteries. Area under the vasodilatation curve (AUC) calculated for each response to (a) flow and (b) acetylcholine in the absence or presence of ADMA (1 mM). Results are mean±s.e.m. of eight to nine preparations. Differences in responses evaluated by a two-way analysis of variance: *P<0.05 versus control response.
Effect of blockers of Ca2+-activated K+ channels and herbimycin on flow-evoked vasodilatation
In the presence of indomethacin, incubation with the combination of inhibitors of small-conductance Ca2+-activated K+ channels, apamin (0.5 μM), and intermediate- and largeconductance Ca2+-activated K+ channels, ChTx (0.1 μM) did not change flow- or acetylcholine-evoked vasodilatation (Figure 5a,b).
Figure 5.

The combination of the blockers of Ca2+-activated K+ channels, apamin and ChTx, abolished acetylcholine, but not flow-evoked vasodilatation persisting in the presence of indomethacin and ADMA. Area under the vasodilatation curve (AUC) for (a, c) flow and (b, d) acetylcholine in the absence or presence of a combination of a blocker of small-conductance, apamin (0.5 μM), and intermediate- and large-conductance, ChTx (0.1 μM) Ca2+-activated K+ channels. The experiments were performed in (a, b) the presence of indomethacin (3 μM) and (c, d) the presence of indomethacin and ADMA (1 mM). Results are mean±s.e.m. of four to five preparations. Differences in responses evaluated by two-way analysis of variance: *P<0.05 versus responses obtained in the presence of indomethacin and ADMA.
In the presence of indomethacin and ADMA, incubation with the combination of apamin (0.5 μM) and ChTx (0.1 μM) increased the vasoconstriction induced by U46619 (0.1 μM) from 198 ±13 to 182 ±13 μM (n=5). However, both low and high shear stress levels caused vasodilatation to similar levels in the absence and the presence of apamin and ChTx (Figure 5c). In contrast, in the presence of indomethacin and ADMA, the combination of apamin and ChTx abolished acetylcholine-evoked vasodilatation (Figure 5d). Incubation with a blocker of inward rectifier K+ channels, BaCl2 (100 μM), did not change vasodilatations evoked by flow or acetylcholine (n=3, results not shown).
In the presence of indomethacin, an inhibitor of the tyrosine kinase c-Src, herbimycin A, had a pronounced effect on flow-induced vasodilatation (Figure 6a). Thus, herbimycin A reduced low and high flow-evoked relaxation by, respectively, 32±12 and 68±14% (P<0.05, n=7). In contrast, acetylcholine-evoked vasodilatation in the same vascular preparations was not affected in the presence of herbimycin A (Figure 6b). In the presence of indomethacin and ADMA, incubation with herbimycin A did not cause additional inhibition of flow-evoked vasodilatation, and acetylcholine vasodilatation remained unchanged (Figure 6c, d). In segments treated with indomethacin, incubation with another tyrosine kinase inhibitor, genistein (5 μM), lowered U46619-induced constriction with 50%, but genistein did not affect flow-evoked vasodilatation in rat mesenteric arteries (n=2, results not shown). In rat mesenteric small arteries constricted with U46619, a putative activator of tyrosine kinase, phenylarsine oxide (30 μM), abolished tone irreversibly and therefore, this pathway could not be explored further (n=3).
Figure 6.

The tyrosine kinase inhibitor, herbimycin A, inhibits the sustained vasodilatation by flow, but it does not change acetylcholine vasodilatation. Area under the vasodilatation curve (AUC) for (a, c) flow and (b, d) acetylcholine in the absence or the presence of a tyrosine kinase (c-SRC) inhibitor, herbimycin A (1 μM). The experiments were performed in (a, b) the presence of indomethacin (3 μM) and (c, d) the presence of indomethacin and ADMA (1 mM). Results are mean±s.e.m. of seven to nine preparations. Differences in responses evaluated by two-way analysis of variance: *P<0.05 versus responses obtained in the presence of indomethacin.
Discussion
The main findings of the present study are that flow-evoked vasodilatation was markedly reduced in the presence of an inhibitor of NO synthase and of Src kinase, while these treatments did not cause major changes in acetylcholine-evoked vasodilatation. Moreover, the combination of the K+ channel blockers, apamin and ChTx, inhibited acetylcholine vasodilatation persisting in the presence of inhibitors of cyclooxygenase and NOS. Therefore, these results suggest the endothelial cell signal transduction as well as the contribution of endothelium-derived NO and EDHF-type vasodilatation is different for flow- and acetylcholine-evoked vasodilatation in rat mesenteric small arteries.
Role of endothelium and NO in flow- and acetylcholine-evoked vasodilatation
The range of shear stress applied in the present study corresponds to that in vivo, and maximal vasodilatation should be obtained at these shear stress levels (Griffith, 2000). However, the magnitude of vasodilatation in the present study was less than observed in other studies of mesenteric small arteries, where flow-evoked vasodilatation was elicited on spontaneous tone (Matrougui et al., 1997; Izzard & Heagerty, 1999) or constrictions induced by α1-adrenoceptor antagonists (Takamura et al., 1999). Similar to other studies from this laboratory (Chlopicki et al., 2001), arterial segments that are second to third-order branches of the superior mesenteric artery do not develop spontaneous myogenic tone, and in the case of constrictions elicited by α1-adrenoceptor agonists, washout contributed to the vasodilatation evoked by the flow levels applied in the present study. Therefore, the thromboxane analog, U46619, was applied in the present study and this could be a limitation, since the processes leading to vasoconstriction evoked by the contractile drugs seem to play a role for relaxations mediated by endothelium-derived relaxing factors (Plane & Garland, 1996; Tomioka et al., 1999). Thus, U46619 was suggested to cause a marked inhibition of acetylcholine relaxation in rat mesenteric small arteries in the presence of NOS blockade (Plane & Garland, 1996). However, in a previous study (Prieto et al., 1998) and also in the present study, we applied lower concentrations of U46619 and obtained maximal vasodilatation to acetylcholine. Moreover, the constriction of the arteries by U46619 to 45% of the passive diameter of the arteries allowed large changes in the resistance of the vascular segments when vasodilatation was evoked by flow.
Vasodilatation by elevation in shear stress following increased flow is considered to be endothelium dependent. There are only few in vivo studies which have addressed this issue, but in vitro studies have repeatedly shown that flow-induced vasodilatation is either partially inhibited (Pourageaud & Freslon, 1995; Vequaud et al., 1999) or abolished by endothelial cell removal in small arteries (Izzard & Heagerty, 1999; Takamura et al., 1999). In the present study, a small vasodilatation to flow persisted after removal of the endothelial cell layer and can probably be ascribed to the small pressure variation generated by the pump suction in our setup, which on the other hand allowed us to control precisely the generated flow. However, mechanical removal of the endothelium abolished acetylcholine-evoked vasodilatation and markedly reduced flow-induced vasodilatation indicating that both responses are dependent on an intact endothelial cell layer.
NO plays a main role for both agonist and flow-evoked endothelium-dependent vasorelaxation in large arteries (Cooke et al., 1991; Simonsen et al., 1999; Danser et al., 2000). In human small arteries and rat skeletal arterioles, prostanoids and NO were found only to have a minor role in acetylcholine-evoked vasodilatation (Buus et al., 2000; Ungvari et al., 2001), while flow-evoked vasodilatation was abolished in the presence of inhibitors of cyclooxygenase and NO (Paniagua et al., 2001). The same preconstrictor noradrenaline was applied and therefore, these studies suggest that different activation of the endothelial cell layer is associated with the release of different types of endothelium-derived relaxant factor. In rat mesenteric small arteries with spontaneous tone, inhibition of NO synthase and cyclooxygenase was demonstrated either to cause no or partial inhibition of acetylcholine and flow-evoked vasodilatation (Iglarz et al., 1998; Izzard & Heagerty, 1999; Takamura et al., 1999). In the present study, the arteries were constricted with the thromboxane analog, U46619, and an inhibitor of NO synthase, ADMA, caused pronounced inhibition of flow-evoked vasodilatation. The concentration of ADMA, applied in the present study, causes effective inhibition of NO synthase, since it reduces acetylcholine-evoked NO release in the rat superior mesenteric artery to levels similar to that of NG-nitro-L-arginine (Simonsen et al., 1999; Stankevicius et al., 2002). Therefore, the main part of shear stress-induced vasodilatation of mesenteric small arteries can be attributed to NO, while NO only seems to play a minor role for acetylcholine-evoked vasodilatation.
Role of nonprostanoid non-NO in flow- and acetylcholine-evoked vasodilatation
Endothelial cell calcium plays a pivotal role for agonist-induced release of endothelium-derived relaxing factors including prostacyclin, NO, and EDHF-type vasorelaxation (Nilius & Droogmans, 2001). Simultaneous recordings of endothelial membrane potential and cytosolic Ca2+ in the intact rat aorta also showed that acetylcholine-evoked hyperpolarization coincides with rises in Ca2+, suggesting these two events are coupled (Carter & Ogden, 1994; Usachev et al., 1995; Nilius & Droogmans, 2001). Patch-clamp experiments and reverse transcriptase–polymerase chain reaction (RT–PCR) have also provided evidence for the presence of small, intermediate, and large-conductance Ca2+-activated K+ channels in endothelial cells of intact arteries (Marchenko & Sage, 1996; Kohler et al., 2000), while small- and intermediate-conductance Ca2+-activated K+ channels are not expressed in mature smooth muscle cells (Burnham et al., 2002; Bychkov et al., 2002). Moreover, measurement of endothelial cell membrane potential in intact mesenteric small arteries suggested endothelial small- and intermediate-conductance Ca2+-activated K+ channels to be involved in acetylcholine-induced EDHF-type relaxation (Edwards et al., 1998). Therefore, the present study confirms previous studies (Edwards et al., 1998; Buus et al., 2000; Busse et al., 2002), where the blockade of Ca2+-activated K+ channels with the combination of apamin and ChTx was also found to inhibit acetylcholine vasodilatation resistant to cyclooxygenase and NOS inhibition.
Shear stress-induced vasodilatation is only in part dependent on rises in endothelial cell calcium (Muller et al., 1999; Busse & Fleming, 2003), but Ca2+-activated K+ channels have been suggested to be involved in flow-evoked vasodilatation. Vasodilatation of large arteries, evoked by superfusate either from cultured endothelial cells exposed to flow or from a perfused endothelium-intact donor artery, was inhibited in the presence of a blocker of Ca2+-activated K+ channels, tetraethylammonium (Cooke et al., 1991; Hutcheson & Griffith, 1994). Moreover, tetraethylammonium and ChTx was found to inhibit the non-NO nonprostanoid vasodilatation and hyperpolarization evoked by flow in coronary arterioles (Dube & Canty, 2001; Miura et al., 2001). An inhibitor of large-conductance Ca2+-activated K+ channels, iberiotoxin, was by intraluminal application also found to inhibit flow-dependent dilation in rat mesenteric arterioles, while the effect of the combination of apamin and ChTx on flow was small in medium-sized mesenteric arteries (Takamura et al., 1999). Therefore, arterial size, the vascular bed studied, species differences, and probably also experimental conditions seem to play a role for the implication of Ca2+-activated K+ channels in flow-evoked vasodilatation. In the present study, we compared in the same preparations the effect of the combination of apamin and ChTx on acetylcholine- and flow-evoked vasodilatation and found that in contrast to acetylcholine-evoked vasodilatation, flow-evoked vasodilatation was not sensitive to the inhibition of Ca2+-activated K+ channels. These results suggest that opening of Ca2+-activated K+ channels does not appear to contribute to shear stress-evoked vasodilatation of U46619-constricted rat mesenteric small arteries.
Shear stress enhances the open probability of inward rectifying K+ channels in cellattached patches (Olesen et al., 1988; Nilius & Droogmans, 2001), and a blocker of these channels, BaCl2, was reported to inhibit flow-evoked vasodilatation in the rabbit iliac arteries (Cooke et al., 1991). However, in the present study a lower concentration (100 μM) of BaCl2, which only inhibits the inward rectifying K+ channels, did not reduce flow-evoked vasodilatation. Therefore, opening of BaCl2-sensitive inward rectifier and Ca2+-activated K+ channels does not appear to contribute to shear stress-evoked vasodilatation of U46619-constricted rat mesenteric small arteries, although it cannot be excluded that application of even higher levels of shear stress than applied in the present study will lead to opening of endothelial cell K+ channels.
Calcium-independent mechanisms are involved in the formation of endothelium-derived relaxing factors. Thus, tyrosine kinase and serine/threonine kinases were found to be involved in phosphorylation and activation of endothelial NOS (Corson et al., 1996; Fleming et al., 1998; Dimmeler et al., 1999). Tyrosine kinases sensitive to genistein were found to be involved in endothelial NOS activation by flow in rat coronary (Muller et al., 1996) and gracialis muscle arterioles (Ungvari et al., 2001). However, in isolated human umbilical vein endothelial cells and porcine aortic endothelial cells shear stress-evoked phosphorylation of extracellular signal-regulated kinase (ERK1/2) and endothelial NOS was found to be dependent on genistein-insensitive tyrosine kinase, which was inhibitedin the presence of herbimycin (Takahashi & Berk, 1996; Fleming et al., 1998). Herbimycin is a tyrosine kinase with Src selectivity, and in the present study herbimycin inhibited shear stress-induced vasodilatation without alteration in acetylcholine-evoked vasodilatation. These observations suggest that the flow-evoked, mainly NO-mediated, vasodilatation, in contrast to acetylcholine vasodilatation, depends on the activation of Src tyrosine kinase in rat mesenteric arteries.
The present study suggests that Ca2+-activated K+ channels sensitive for the combination of apamin and ChTx are involved in acetylcholine-evoked, mainly EDHF-type, vasodilatation, while a Src tyrosine kinase plays a role for flow-evoked NO-mediated vasodilatation in rat mesenteric small arteries.
Acknowledgments
This work was supported by grants from the Danish Medical Research Council, the Danish Heart Foundation 00-2-1-10-22834, and the Research Foundation of the University of Aarhus.
Abbreviations
- ADMA
NG,NG-asymmetric dimethyl-L-arginine
- AUC
area under curve
- BaCl2
barium chloride
- ChTx
charybdotoxin
- EDHF
endothelium-derived hyperpolarizing factor
- EDTA
ethylenediaminetetraacetic acid
- NO
nitric oxide
- NOS
nitric oxide synthase
- PSS
physiological salt solution
- U46619
9,11-di-deoxy-11α, 9α-epoxymethano prostaglandin F2α
References
- BAUERSACHS J., HECKER M., BUSSE R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid meta-bolite in the coronary microcirculation. Br. J. Pharmacol. 1994;113:1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLZ S.S., FISSLTHALER B., PIEPERHOFF S., DE WIT C., FLEMING I., BUSSE R., POHL U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca(2+) changes and dilation in isolated resistance arteries. FASEB J. 2000;14:255–260. doi: 10.1096/fasebj.14.2.255. [DOI] [PubMed] [Google Scholar]
- BURNHAM ,M.P., BYCHKOV R., FELETOU M., RICHARDS G.R., VANHOUTTE P.M., WESTON A.H., EDWARDS G. Characterization of an apamin-sensitive small-conductance Ca(2+)- activated K(+) channel in porcine coronary artery endothelium: relevance to EDHF. Br. J. Pharmacol. 2002;135:1133–1143. doi: 10.1038/sj.bjp.0704551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSSE R., EDWARDS G., FELETOU M., FLEMING I., VANHOUTTE P.M., WESTON A.H. EDHF: bringing the concepts together. Trends Pharmacol. Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- BUSSE R., FLEMING I. Regulation of endothelium-derived vasoactive autocoid production by hemodynamic forces. Trends Pharmacol. Sci. 2003;24:24–29. doi: 10.1016/s0165-6147(02)00005-6. [DOI] [PubMed] [Google Scholar]
- BYCHKOV R., BURNHAM M.P., RICHARDS G.R., EDWARDS G., WESTON A.H., FELETOU M., VANHOUTTE P.M. Characterization of a charybdotoxin sensitive intermediate conductance Ca(2+)-activated K(+) channel in porcine coronary endothelium: relevance to EDHF. Br. J. Pharmacol. 2002;137:1346–1354. doi: 10.1038/sj.bjp.0705057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUUS N.H., SIMONSEN U., PILEGAARD H.K., MULVANY M.J. Nitric oxide, prostanoid and non-NO, non-prostanoid involvement in acetylcholine relaxation of isolated human small arteries. Br. J. Pharmacol. 2000;129:184–192. doi: 10.1038/sj.bjp.0703041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUUS N.H., VANBAVEL E., MULVANY M.J. Differences in sensitivity of rat mesenteric small arteries to agonists when studied as ring preparations or as cannulated preparations. Br. J. Pharmacol. 1994;112:579–587. doi: 10.1111/j.1476-5381.1994.tb13114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARTER T.D., OGDEN D. Acetylcholine-stimulated changes of membrane potential and intracellular Ca2+ concentration recorded in endothelial cells in situ in the isolated rat aorta. Pflugers Arch. 1994;428:476–484. doi: 10.1007/BF00374568. [DOI] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHLOPICKI S., NILSSON H., MULVANY M.J. Initial and sustained phases of myogenic response of rat mesenteric small arteries. Am. J. Physiol. 2001;281:H2176–H2183. doi: 10.1152/ajpheart.2001.281.5.H2176. [DOI] [PubMed] [Google Scholar]
- COLEMAN H.A., TARE M., PARKINGTON H.C. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am. J. Physiol. 2001;280:H2478–H2783. doi: 10.1152/ajpheart.2001.280.6.H2478. [DOI] [PubMed] [Google Scholar]
- COOKE J.P., ROSSITCH E., JR, ANDON N.A., LOSCALZO J., DZAU V.J. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J. Clin. Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORSON M.A., JAMES N.L., LATTA S.E., NEREM R.M., BERK B.C., HARRISON D.G. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ. Res. 1996;79:984–991. doi: 10.1161/01.res.79.5.984. [DOI] [PubMed] [Google Scholar]
- DANSER A.H., TOM B., DE VRIES R., SAXENA P.R. L-NAME-resistant bradykinin-induced relaxation in porcine coronary arteries is NO-dependent: effect of ACE inhibition. Br. J. Pharmacol. 2000;131:195–202. doi: 10.1038/sj.bjp.0703555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMMELER S., FLEMING I., FISSLTHALER B., HERMANN C., BUSSE R., ZEIHER A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- DUBE S., CANTY J.M., JR Shear stress-induced vasodilatation in porcine coronary conduit arteries is independent of nitric oxide release. Am. J. Physiol. 2001;280:H2581–H2590. doi: 10.1152/ajpheart.2001.280.6.H2581. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FLEMING I., BAUERSACHS J., FISSLTHALER B., BUSSE R. Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ. Res. 1998;82:686–695. doi: 10.1161/01.res.82.6.686. [DOI] [PubMed] [Google Scholar]
- FULTON D., MCGIFF J.C., QUILLEY J. Contribution of NO and cytochrome P450 to the vasodilator effect of bradykinin in the rat kidney. Br. J. Pharmacol. 1992;107:722–725. doi: 10.1111/j.1476-5381.1992.tb14513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIFFITH T.M. A nitric oxide. Biology and Pathophysiology 2000San Diego: Academic Press; 570–595.ed. Ignarro, L.J. pp [Google Scholar]
- HUTCHESON I.R., GRIFFITH T.M. Heterogeneous populations of K+ channels mediate EDRF release to flow but not agonists in rabbit aorta. Am. J. Physiol. 1994;266:H590–H596. doi: 10.1152/ajpheart.1994.266.2.H590. [DOI] [PubMed] [Google Scholar]
- HWA J.J., GHIBAUDI L., WILLIAMS P., CHATTERJEE M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am. J. Physiol. 1994;266:H952–H958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- IGLARZ M., MATROUGUI K., LEVY B.I., HENRION D. Chronic blockade of endothelin ETA receptors improves flow dependent dilation in resistance arteries of hypertensive rats. Cardiovasc. Res. 1998;39:657–664. doi: 10.1016/s0008-6363(98)00151-5. [DOI] [PubMed] [Google Scholar]
- IZZARD A.S., HEAGERTY A.M. Impaired flow-dependent dilatation in distal mesenteric arteries from the spontaneously hypertensive rat. J. Physiol. 1999;518:239–245. doi: 10.1111/j.1469-7793.1999.0239r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOHLER R., DEGENHARDT C., KUHN M., RUNKEL N., PAUL M., HOYER J. Expression and function of endothelial Ca(2+)-activated K(+) channels in human mesenteric artery: a single-cell reverse transcriptase–polymerase chain reaction and electrophysiological study in situ. Circ. Res. 2000;87:496–503. doi: 10.1161/01.res.87.6.496. [DOI] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATOBA T., SHIMOKAWA H., NAKASHIMA M., HIRAKAWA Y., MUKAI Y., HIRANO K., KANAIDE H, TAKESSHITA A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Invest. 2000;106:1521–1531. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATROUGUI K., MACLOUF J., LEVY B.I., HENRION D. Impaired nitric oxide- and prostaglandin-mediated responses to flow in resistance arteries of hypertensive rats. Hypertension. 1997;30:942–947. doi: 10.1161/01.hyp.30.4.942. [DOI] [PubMed] [Google Scholar]
- MCGUIRE J.J., DING H., TRIGGLE C.R. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s) Can. J. Physiol. Pharmacol. 2001;79:443–470. [PubMed] [Google Scholar]
- MIURA H., WACHTEL R.E., LIU Y., LOBERIZA F.R., JR, SAITO T., MIURA M., GUTTERMAN D.D. Flow-induced dilation of human coronary arterioles: important role of Ca(2+)-activated K(+) channels. Circulation. 2001;103:1992–1998. doi: 10.1161/01.cir.103.15.1992. [DOI] [PubMed] [Google Scholar]
- MULLER J.M., DAVIS M.J., CHILIAN W.M. Coronary arteriolar flow-induced vasodilatation signals through tyrosine kinase. Am. J. Physiol. 1996;270:H1878–H1884. doi: 10.1152/ajpheart.1996.270.6.H1878. [DOI] [PubMed] [Google Scholar]
- MULLER J.M., DAVIS M.J., KUO L., CHILIAN W.M. Changes in coronary endothelial cell Ca2+ concentration during shear stress- and agonist-induced vasodilatation. Am. J. Physiol. 1999;276:H1706–H1714. doi: 10.1152/ajpheart.1999.276.5.H1706. [DOI] [PubMed] [Google Scholar]
- NILIUS B., DROOGMANS G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- OLESEN S.P., CLAPHAM D.E., DAVIES P.F. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- PANIAGUA O.A., BRYANT M.B., PANZA J.A. Role of endothelial nitric oxide in shear stress-induced vasodilatation of human microvasculature: diminished activity in hypertensive and hypercholesterolemic patients. Circulation. 2001;103:1752–1758. doi: 10.1161/01.cir.103.13.1752. [DOI] [PubMed] [Google Scholar]
- PARSONS S.J., HILL A., WALDRON G.J., PLANE F., GARLAND C.J. The relative importance of nitric oxide and nitric oxide-independent mechanisms in acetylcholine-evoked dilatation of the rat mesenteric bed. Br. J. Pharmacol. 1994;113:1275–1280. doi: 10.1111/j.1476-5381.1994.tb17136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., GARLAND C.J. Influence of contractile agonists on the mechanism of endothelium-dependent relaxation in rat isolated mesenteric artery. Br. J. Pharmacol. 1996;119:191–193. doi: 10.1111/j.1476-5381.1996.tb15970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POURAGEAUD F., FRESLON J.L. Impaired endothelial relaxations induced by agonists and flow in spontaneously hypertensive rat compared to Wistar–Kyoto rat perfused coronary arteries. J. Vasc. Res. 1995;32:190–199. doi: 10.1159/000159093. [DOI] [PubMed] [Google Scholar]
- PRIETO D., SIMONSEN U., HERNANDEZ M., GARCIA-SACRISTAN A. Contribution of K+ channels and ouabain-sensitive mechanisms to the endothelium-dependent relaxations of horse penile small arteries. Br. J. Pharmacol. 1998;123:1609–1620. doi: 10.1038/sj.bjp.0701780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUBANYI G.M., ROMERO J.C., VANHOUTTE P.M. Flow-induced release of endothelium-derived relaxing factor. Am. J. Physiol. 1986;252:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- SANDOW S.L., HILL C.E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- SHIMOKAWA H., YASUTAKE H., FUJII K., OWADA M.K., NAKAIKE R., FUKUMOTO Y., TAKAYANAGI T., NAGAO T., EGASHIRA K., FUJISHIMA M., TAKESHITA A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- SIMONSEN U., WADSWORTH R.M., BUUS N.H., MULVANY M.J. In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. J. Physiol. 1999;516:271–282. doi: 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANKEVICIUS E., MARTINEZ A.C., MULVANY M.J., SIMONSEN U. Blunted acetylcholine relaxation and nitric oxide release in arteries from renal hypertensive rats. J. Hyperten. 2002;20:1571–1579. doi: 10.1097/00004872-200208000-00020. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI M., BERK B.C. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. Essential role for a herbimycin-sensitive kinase. J. Clin. Invest. 1996;98:2623–2631. doi: 10.1172/JCI119083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAMURA Y., SHIMOKAWA H., ZHAO H., IGARASHI H., EGASHIRA K., TAKESHITA A. Important role of endothelium-derived hyperpolarizing factor in shear stress-induced endothelium-dependent relaxations in the rat mesenteric artery. J. Cardiovasc. Pharmacol. 1999;34:381–387. doi: 10.1097/00005344-199909000-00010. [DOI] [PubMed] [Google Scholar]
- TAYLOR H.J., CHAYTOR A.T., EDWARDS D.H., GRIFFITH T.M. Gap junction-dependent increases in smooth muscle cAMP underpin the EDHF phenomenon in rabbit arteries. Biochem. Biophys. Res. Commun. 2001;283:583–589. doi: 10.1006/bbrc.2001.4791. [DOI] [PubMed] [Google Scholar]
- TOMIOKA H., HATTORI Y., FUKAO M., SATO A., LIU M., SAKUMA I., KITABATAKE A., KANNO M. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: importance of processes mediating precontractions. J. Vasc. Res. 1999;36:311–320. doi: 10.1159/000025659. [DOI] [PubMed] [Google Scholar]
- UNGVARI Z., SUN D., HUANG A., KALEY G., KOLLER A. Role of endothelial [Ca2+]i in activation of eNOS in pressurized arterioles by agonists and wall shear stress. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H606–H612. doi: 10.1152/ajpheart.2001.281.2.H606. [DOI] [PubMed] [Google Scholar]
- USACHEV Y.M., MARCHENKO S.M., SAGE S.O. Cytosolic calcium concentration in resting and stimulated endothelium of excised intact rat aorta. J. Physiol. 1995;489:309–317. doi: 10.1113/jphysiol.1995.sp021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEQUAUD P., POURAGEAUD F., FRESLON J.L. Role of nitric oxide and endothelium in the flow-induced dilation of rat coronary arteries under two preconstriction conditions. Clin. Exp. Pharmacol. Physiol. 1999;26:470–476. doi: 10.1046/j.1440-1681.1999.03061.x. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO Y., IMAEDA K., SUZUKI H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTATT E.D. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]