

Abstract
The kinetics of charge transfer depend crucially on the dielectric reorganization of the medium. In enzymatic reactions that involve charge transfer, atomic dielectric response of the active site and of its surroundings determines the efficiency of the protein as a catalyst. We report direct spectroscopic measurements of the reorganization energy associated with the dielectric response in the active site of α-chymotrypsin. A chromophoric inhibitor of the enzyme is used as a spectroscopic probe. We find that water strongly affects the dielectric reorganization in the active site of the enzyme in solution. The reorganization energy of the protein matrix in the vicinity of the active site is similar to that of low-polarity solvents. Surprisingly, water exhibits an anomalously high dielectric response that cannot be described in terms of the dielectric continuum theory. As a result, sequestering the active site from the aqueous environment inside low-dielectric enzyme body dramatically reduces the dielectric reorganization. This reduction is particularly important for controlling the rate of enzymatic reactions.
Many enzymatic reactions are charge transfer processes. Their activation free energy is affected by the dielectric properties of the milieu of the reacting groups (1). It has been suggested that small dielectric response at the active site could be a physical reason for high catalytic activity (2–4).
Direct measurements of local dielectric properties at enzyme active sites have never been made. Most of the current knowledge is based on circumstantial evidence. Different existing data can be used to support opposing points of view. For instance, the average static dielectric constant of proteins ɛs ≈ 4 was estimated from measurements on dry powders (5–9). However, x-ray data indicate that the mobility of atoms near enzymes active sites is higher than average (10, 11) and, thus, the dielectric response should be enhanced. Furthermore, active sites are typically situated within several angstroms of the protein surface. A wide range of dielectric response was predicted from molecular simulations for various proteins: A gradual increase from ɛ ≈ 3 in the center of globule to about ɛ ≈ 10 at its periphery was found in refs. 12–19. Even higher values of 25–35 were calculated by taking into account fluctuations of ionizable surface groups (18, 19). Finally, highly polarizable aqueous surroundings may contribute to the dielectric response, to increase the uncertainty even further.
Thus, local dielectric properties at enzyme active sites have remained unclear. In the present paper, we attempt to resolve this issue by direct measurement. A spectroscopic technique proposed in refs. 20 and 21 and further developed in refs. 22–24 is used. The reorganization energy is determined from absorption and emission spectra of proflavine noncovalently bound in the active site of α-chymotrypsin. We evaluate the contributions to the dielectric reorganization of the protein interior and of the surrounding solvent by comparing the spectra of the dye–enzyme complex in a solid protein film and in an aqueous solution.
Methods
Theory.
The activation free energy of charge transfer, ΔG≠, is determined by the reaction free energy, ΔG, by the energetic cost of distortion of chemical bonds in reacting groups, λi, and by the energetic cost of repolarization (reorganization) of the surrounding medium, λs, (1)
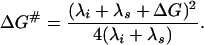 |
1 |
The focus of the present work is the reorganization free energy of the medium, λs, which is determined by the atomic (inertial) dielectric response to charge transfer.
When a chromophore undergoes an optical electronic transition accompanied by absorption or emission of light, the corresponding change in its electron density distribution causes repolarization of the surrounding medium. As a result, the medium reorganization energy affects the electronic spectra. As illustrated in Fig. 1, this effect can be used for spectroscopic determination of λs (20, 21). For a classical system, the difference in the absorption νa and emission νe maxima of the chromophore (the Stokes shift) is given by hνa − hνe = 2(λs + λi), where h is the Planck's constant. Quantum effects produce an important correction (22) that, for the case of featureless spectral bands, is
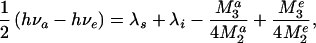 |
2 |
where M2a, M2e, M3a, and M3e are, respectively, the second and third central moments of the absorption and emission spectra. The values of νa, νe, M2, and M3 can be determined from each experimental spectrum. Then, if the intramolecular reorganization energy, λi, for the chromophore is known, the value of λs for the medium can be directly extracted with the help of Eq. 2.
Figure 1.

A schematic representation of the energetics of electronic transitions in a dye in a polarizable medium (Left) and the corresponding absorption and emission spectra for proflavine dye in a polar medium (Right). The x axis on the left is a generalized classical coordinate describing, e.g., the orientation of surrounding dipoles in the medium. The light absorption (hνa) and emission (hνe) transitions are shown at the most probable coordinates that are expected to determine the maxima of the corresponding spectra.
The intramolecular reorganization energy for proflavine, has been previously determined by comparing the measured λs + λi with calculated λs for a series of solvents with well characterized dielectric properties (24). The medium reorganization energy, denoted here as λscal, was calculated in ref. 24 within the framework of the dielectric continuum model as follows. The cavity formed by proflavine in the surrounding solvent was approximated by an oblate spheroid. The electron density redistribution upon the optical transition, found from semiempirical quantum chemical calculations, was placed inside the cavity. The potential created by this charge density was determined from an analytical solution of the Poisson equation with macroscopic values of the optical and static dielectric constants of the solvent. Intrinsic uncertainties in the shape of the cavity and in the electron density redistribution were accounted for by a scaling parameter p, assuming that the true value of λs is related to λscal as λs = p λscal. The values of λi and p were extracted simultaneously by linear regression analysis of the dependence of measured λs + λi on calculated λscal for a series of reference solvents (24).
In the present work, we reanalyzed the data of ref. 24, using the van der Waals surface of proflavine rather than an oblate spheroid as the solvent-inaccessible cavity, and solving the Poisson equation numerically with the help of the delphi program (25). This geometry reduced the uncertainties and resulted in p ≈ 2 compared with p ≈ 4 obtained in ref. 24. The value of λi ≈ 455 cm−1 remained unchanged. In addition, we corroborated semiempirical quantum chemical calculations of the charge redistribution in the monocationic proflavine by 6-31G** ab initio calculations that gave similar results.
Materials.
Proflavine hemisulfate was purchased from Fluka, and bovine α-chymotrypsin (dialyzed, essentially salt free, Type II), and other chemicals were purchased from Sigma.
Proflavine–Chymotrypsin Complex.
Preparation of the complex was as described in ref. 26. In brief, solutions containing 0.5–4 mM chymotrypsin, 1–10 μM proflavine, 0–0.5 M KCl, and 10 mM Tris/10 mM citrate (pH 8.0–8.5) at 20°C produced complexes of the dye with the enzyme unprotonated at His57 in the active site. Solutions of the protonated complex were prepared in 0.5 M KCl and 10 mM Tris/10 mM citrate (pH 6.2). Under these conditions, 99.0–99.9% of dye molecules were bound (26).
The dye–enzyme complex was also embedded into solid chymotrypsin films by drying salt-free complex solutions on the internal side of a 10-mm spectroscopic quartz cell. The solutions (pH 8.0–8.5) contained 4–8 mM chymotrypsin and ≈200 μM or ≈0.3 μM proflavine for absorption or emission experiments, respectively. Virtually all dye molecules were bound before drying because of the high enzyme/dye ratio (>20). Initially, the films were dried under the flow of gaseous nitrogen at room temperature. For further drying, a small open container with a saturated solution of KNO3, KCl, K2CO3, or with concentrated H2SO4 was placed inside the sealed cell to keep constant humidity. After 20–50 hours of equilibration, these cells were used for spectroscopic measurements. On films, absorption and fluorescence measurements were made by using samples that were ≈10 μm (≈3,000 molecular layers) thick (estimated from light absorption by the dye, and from the film weight and density assumed to be ≈1 g/cm3). For circular dichroism measurements, ≈200 molecular-layer-thick films were made.
Chloroform infusion was performed by dropping 30 μl of chloroform onto a film of ≈6-mg weight. After this, the cell was resealed with the same saturated salt solution container inside and was stored for 60–90 min before spectroscopic measurements. The film located on the vertical wall of the cell remained soaked in chloroform whereas residual solvent stayed at the cell bottom. After equilibration of a film submerged in excess of chloroform for several hours, no traces of dye extraction into chloroform were found.
Spectroscopic Measurements.
In all experiments, proflavine was present in the monocationic form, as judged from its spectra. Absorption spectra of proflavine in water and in the enzyme–dye complex were recorded in 10-mm quartz cells on a Perkin–Elmer Lambda 40P spectrophotometer at 1-nm slit width, 200-nm/min scanning speed, and 1-s time constant. The optical density at the absorption maximum of the dye was 0.1–0.5. In salt- and protein-free aqueous solutions, the dye concentration was <4 μM to prevent aggregation. To obtain the dye spectrum in a dye–enzyme complex, the background spectrum of the same sample containing no dye was subtracted from the sample spectrum.
Fluorescence spectra were recorded in 10- × 10-mm quartz cells on a Hitachi 850 spectrofluorometer at 5-nm emission slit width, 2-nm excitation slit width, 60-nm/min scanning speed, and 2-s time response. The optical density of the solutions at the absorption maximum of the dye was less than 0.15. The standard correction (27) for the instrument response by using Rhodamin B quantum counter and light diffuser was performed. To resolve scattered excitation beam and emitted light, the spectra of protein films were recorded in a solid sample holder with the sample plane tilted with respect to the excitation beam, and the 440-nm excitation wavelength was used. The blue shift of the excitation with respect to the absorption maximum (460–466 nm) had virtually no effect on the shape of the spectra.
We estimated solvent accessibility of proflavine in the dye–enzyme complex by comparing fluorescence quenching by iodide ions for the free dye in water (pH 6) and for the complex in solution at pH 8.5. The quenching constant was determined as described in ref. 28 from the dependence of the intensity at the emission maximum on [I−]/[Cl−] ratio at fixed 0.05 M ionic strength in KCl/KI solutions.
A similar procedure was used to control the lifetime of the excited state for evaluation of the role of slow dielectric relaxation in the overall dielectric response. The relative change in the lifetime upon variation in [I−]/[Cl−] ratio at fixed 0.5 M ionic strength (pH 8.5) was evaluated from the quantum yield monitored via the intensity at the emission maximum. The characteristic time and amplitude of dielectric relaxation in the complex was extracted from the dependence of the emission maximum on the lifetime as described in refs. 28 and 29.
Circular dichroism spectra of chymotrypsin in solution (7 μM chymotrypsin/2 mM Tris, pH 8.5; 1-mm quartz cells) and in solid protein films were recorded on a Jasco 715 spectrometer at 2-nm slit width, 20-nm/min scanning speed, and 2-s response time. Activity of chymotrypsin was determined from the initial rate of hydrolysis of N-acetyl-l-tyrosine ethyl ester monitored by absorbance at 237 nm as described in ref. 30.
Results
We previously found that dielectric reorganization in simple solvents caused by the long-wave optical electronic transition in proflavine is well described within a dielectric continuum model (24). The Stokes shifts determined in ref. 24 for a set of aprotic solvents are plotted in Fig. 2 vs. the calculated reorganization energy (λs). The dashed line in Fig. 2, a fit of this dependence to Eq. 2 obtained in ref. 24, is used as a calibration curve for semiquantitative evaluation of λs in water and in the dye–enzyme complex. This evaluation is possible because the Stokes shift is a monotonic and only weakly nonlinear function of λs (Fig. 2; refs. 22 and 24).
Figure 2.

Semiquantitative evaluation of medium reorganization energy, λs, from the measured Stokes shift of proflavine: ○, calibration data for polar aprotic solvents with known dielectric properties (24) vs. calculated, scaled λs; ♦, the Stokes shift in water vs. λs calculated at ɛs = 80 and ɛo = 1.77. The dashed line shows the fit to Eq. 2 of the data for aprotic solvents (24). This fit can be used as a calibration curve to estimate λs at the active site of chymotrypsin from the measured Stokes shifts of the dye–enzyme complex (shown by arrows). The lowest and uppermost open circles are, respectively, for dichloromethane and acetonitrile. Quantitative evaluation of λs with correction for the spectral moments is described in the text.
Alternatively, we use Eq. 2 to correct the Stokes shift data for measured spectral moments and λi determined from the reference solvents data for more accurate calculation of λs. This method relies on the validity of assumptions underlying Eq. 2 (22, 24). Because the main conclusions of this work can be drawn from such quantitative analysis or directly from the Stokes shift data, they are independent of a particular model. Values of λs presented in the text were obtained by the quantitative method.
Free Dye and Dye–Enzyme Complex in Solution.
For free proflavine in water, the measured Stokes shift is 1,725 cm−1 (Fig. 2); the corresponding λs is ≈1,950 cm−1. A dramatic decrease in the Stokes shift (to 1,075 cm−1) and in λs (to 1,280 cm−1) is observed upon proflavine–chymotrypsin complex formation in aqueous solution (Fig. 2). This change is caused by binding of the dye in the substrate-binding pocket of the enzyme. The latter value of λs represents the reorganization energy at the active site of solvated chymotrypsin. This interpretation is based on the following arguments and control experiments.
Proflavine is a competitive inhibitor of α-chymotrypsin. Experimental studies suggest that the dye binds at a single site in the vicinity of the active center (26, 31, 32). The model shown in Fig. 3 illustrates a possible structure of the complex with the dye in the substrate binding pocket. This structure is similar to the x-ray structure (33) of a complex of proflavine with trypsin, a closely related serine proteinase. The modeling shows that more than three-fourths of the dye surface is shielded from bulk water. We confirmed that by observing a 9-fold drop in the constant of proflavine fluorescence quenching by iodide upon dye–enzyme complex formation.
Figure 3.

The proflavine–chymotrypsin complex. The arrow points to the edge of proflavine molecule. The structure was obtained by 10-ps molecular dynamics simulations. Calculations were performed within hyper chem 5.0 (Hypercube, Gainesville, FL) package using the amber (64) force field with 4,000 water molecules in the periodic box of 56 × 50 × 56 Å with the time step of 1 fs. Partial charges of proflavine were obtained from CNDO/2 semiempirical quantum chemical calculations. Other force field parameters were taken from amber parameters corresponding to similar atoms and chemical bonds of nucleotide bases. X-ray coordinates of chymotrypsin atoms with added hydrogen atoms and equilibrated for 20-ps molecular dynamics run without the dye were taken as the initial configuration of the protein.
We measured proflavine–chymotrypsin complex spectra under such conditions that more than 99% of the dye was bound. We estimated the residence time of the dye in the binding pocket as ≈1 ms, based on the association and dissociation rates determined in ref. 34. Because the residence time is much larger than the lifetime (≈3 ns) of the excited state of the dye, the dye remains bound during the excitation–emission cycle.
The spectra of the complex are independent of ionic strength, at least from 0 to 0.5 M KCl at pH 8.5. Therefore, the ionic atmosphere does not contribute to the measured dielectric reorganization at the active site. The spectra are independent of protein concentration once all dye is bound. If some protein aggregation occurs, it does not affect the results.
Protonation of His57 at the active site of chymotrypsin upon reduction of pH from 8.5 to 6.2 at 0.5 M KCl did not affect the Stokes shift and the reorganization energy. This insensitivity suggests that the intramolecular electric field of chymotrypsin has practically no effect on the charge redistribution in the dye. If this is the case, the calibration curve measured in reference solvents can be used for evaluation of the reorganization energy at the active site.
Finally, slow dielectric reorganization modes may not fully contribute to the Stokes shift because their relaxation time is comparable to or larger than the life time of the exited state of proflavine, but they may still contribute to the true reorganization energy at the active site. To evaluate the role of these modes, we measured a change in the apparent reorganization energy upon variation of the excited state lifetime by dynamic fluorescence quenching following the procedure described in refs. 28 and 29. We also estimated the reorganization energy from the spectral bandwidth as discussed in refs. 22 and 35. This alternative method is less accurate, but it reflects contributions of all slow and fast modes. Both estimates suggested that the effect of the slow relaxation modes is small (≤6% of λs).
Dye–Enzyme Complex in Solid Protein Film.
When the proflavine–chymotrypsin complex is embedded into a solid, dehydrated chymotrypsin film, the Stokes shift decreases to 565 cm−1 (Fig. 2). Note that the inhomogeneous environment of the complex in the film results in broadening of the absorption and emission spectra and, consequently, in a spurious increase in the Stokes shift. Thus, the semiquantitative analysis overestimates λs. Under such conditions, the correction for the spectral moments used in our quantitative method is significant. The resulting λs ≈ 820 cm−1. Another possible artifact, the resonant excitation energy transfer between dye molecules in the inhomogeneous environment, was eliminated by reducing the dye concentration in the film until no further change in the emission spectrum was observed.
The decrease in λs, compared with the solvated proflavine–chymotrypsin complex, is apparently related to the removal of water surrounding the protein. Consistently, the Stokes shift and λs increase with increasing water content (relative humidity) in hydrated films. Infusion of hydrated films with chloroform returns λs to the dry film value (Fig. 4). Chloroform seems to penetrate voids between protein molecules and to displace water from low affinity hydration sites. [It is believed that chymotrypsin has both high and low affinity water-binding sites (36–38)]. Because of its low-polarity, chloroform contributes little to the reorganization energy.
Figure 4.

(a) Stokes shift of proflavine in chymotrypsin films vs. humidity. Lower curve (□) is for films infused with chloroform. (b) Stokes shift in films without chloroform vs. water content. The water content was calculated from humidity by using the water adsorption isotherm on chymotrypsin measured in refs. 36 and 37.
The decrease in λs does not appear to be related to a structural change of the enzyme caused by the removal of water. We confirmed the absence of major structural changes as follows: We recovered more than 90% of the enzyme activity from redissolved films. Chymotrypsin is known to preserve catalytic activity in the solid phase even at 30% humidity (39, 40). Circular dichroism spectra of chymotrypsin (Fig. 5) show only a small apparent difference in the secondary structure of the protein in solution and in a hydrated film (95% humidity). Almost no change in the CD spectrum was observed upon film dehydration.
Figure 5.

Circular dichroism spectra of chymotrypsin in solution and in protein films at fixed humidity.
The observed reorganization energy in the films does not seem to be affected by entrapment of residual salt. Neither addition of 5 Tris molecules per enzyme nor lowering of salt concentration by dialysis before film formation had any effect on the measured spectra.
Discussion
For comparative analysis of the dielectric reorganization in different media, we use continuum–dielectric theory. Within this theory, the reorganization energy, λs, of a uniform medium depends on macroscopic dielectric properties through the optical, ɛo, and static, ɛs, dielectric constants of the medium (1) as
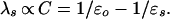 |
3 |
For a dielectrically non-uniform medium, e.g., in the presence of a cavity formed by a chromophoric solute, the relationship between λs and ɛo and ɛs is more complex. Nevertheless, the commonly used parameter C is still a good qualitative descriptor of the reorganizational ability of the medium (23). In addition to λs values calculated by taking into account the dielectric inhomogeneity, we give the corresponding C values.
Dielectric Reorganization of Protein Matrix.
For chymotrypsin embedded in a dehydrated protein film, we find (cf., Fig. 2) that the reorganization energy λs ≈ 820 cm−1 at the active site is similar to that in a low-polarity solvent; e.g., it is smaller than in dichloromethane (ɛo = 2.03, ɛs = 9, C = 0.38). Using this value of λs and assuming that, in a protein film, ɛo ≈ 2.4 ± 0.1, we obtain effective ɛs ≈ 9 ± 1 at the active site, which formally corresponds to effective C ≈ 0.305 ± 0.005. [From the refraction index increment for α-chymotrypsin in solution (41), the specific ɛo for a protein is ≈2.5. We assume that local variations in electron density inside a dehydrated protein film may lower ɛo by ≈10%].
Thus, the dielectric reorganization at the active site of chymotrypsin is stronger than what would be predicted if one were to use ɛs ≈ 4 (C ≈ 0.15) obtained (5–9) for dry protein powders. However, it is weaker than what would be predicted if one were to use ɛs ≈ 20–35 (C ≈ 0.4) calculated in simulations of the dielectric response in proteins (18, 19). The latter large values of ɛs are associated with enhanced mobility of protein groups. In enzymes, the enhanced mobility in the vicinity of the active site may be beneficial for substrate binding but not for the subsequent catalytic charge transfer reaction (42–44). It was found that the mobility in the active site of myoglobin and trypsinogen is reduced after substrate binding (42, 43, 45). This reduction may also occur upon proflavine binding in the active site of chymotrypsin.
Note that small dielectric reorganization in proteins was invoked to explain a variety of kinetic data for charge transfer reactions (1, 3, 4, 46–51) and was inferred from molecular simulations (52–60). Our direct measurements now provide evidence supporting this hypothesis.
Dielectric Reorganization of Water.
For proflavine in water, the dielectric continuum theory predicts λs ≈ 1,450 cm−1, calculated accounting for the dielectric inhomogeneity effect and using ɛo = 1.78 and ɛs = 80 of water (C = 0.55). This predicted λs is close to the maximal λs ≈ 1,500 cm−1 allowed within this theory for this process in common liquids at ambient conditions (ɛo ≥ 1.77, ɛs ≤ ∞, C ≤ 0.56). However, the measured value of the reorganization energy λs ≈ 1,950 cm−1 is anomalously high.
This anomaly is not a result of hydrogen bond formation between the dye and water molecules. Similar hydrogen bonding occurs in alcohols, where it reduces rather than increases the measured Stokes shifts and λs compared with aprotic solvents with similar macroscopic dielectric properties (24). The anomaly is not an artifact of data processing. We reach the same conclusion directly from raw data on the Stokes shift. The value of the Stokes shift calculated from the dielectric continuum theory for a solvent with the same ɛo and ɛs as in water is 1,295 cm−1 vs. the measured 1,725 cm−1 Stokes shift in water (Fig. 2).
The presence of the anomaly suggests that the dielectric continuum theory breaks down at least for the optical short-distance charge transfer process in water. It may be related to the nonlocal dielectric response of water to spatially varying electric fields. We will focus on the physics of the anomaly in a separate work. For the present, it is essential to recognize only that such anomaly can indicate a much larger contribution of water to the reorganization energy of a solvated protein than it can be expected from classical theories.
Effect of Protein Hydration on Reorganization Energy at the Active Site.
To characterize the contribution of water, we measured the change in the reorganization energy of the dye–enzyme complex embedded in a protein film upon variation of water content in the film (Fig. 4 a and b). It is believed that chymotrypsin and other proteins have high- and low-affinity water binding sites (36–38): Below ≈50% relative humidity, water fills primarily high-affinity sites; additional water molecules that hydrate the protein above 50% humidity are weakly bound.
We find that the Stokes shift is virtually unaffected by humidity up to 45% while it increases by ≈160 cm−1 from 45 to 95% humidity (Fig. 4b). This occurs despite the fact that the change in humidity from 0 to 50% brings almost the same amount of water into the protein film as the change in humidity from 50 to 95% (36). Apparently, the mobility of strongly bound water molecules is so low that they contribute to dielectric reorganization far less than weakly bound waters. Similar conclusions can be drawn from the data on the macroscopic dielectric response of water in powders of chymotrypsin (37) and other proteins (61). They are also supported by NMR data showing considerably lower mobility of strongly bound water than the mobility of weakly bound water or bulk water (38, 62, 63).
The reorganization energy at the active site of a fully hydrated dye–enzyme complex in aqueous solution is considerably larger than in a hydrated film at 95% humidity. The presence of bulk water adds another ≈335 cm−1 to the Stokes shift; i.e., it contributes significantly to the reorganization energy at the active site. Most likely the appreciable contribution of bulk water is related to the abnormally large reorganization energy in this anomalous solvent.
Still, λs ≈ 1,280 cm−1 at the active site for the complex in solution is much lower than the reorganization energy for the dye in pure water. For the complex, λs is close to that in polar aprotic solvents (Fig. 2); e.g., it is slightly lower than in acetonitrile (ɛo = 1.8, ɛs = 37, C = 0.53). The difference with pure water is unlikely to be related to a decrease in the number of hydrogen bond-donating groups surrounding the dye at the active site. For instance, removal of such groups upon transfer of the dye from alcohols to polar aprotic solvents results in a decrease in λs (24). The most likely source of the difference is a replacement of the high-reorganization anomalous aqueous solvent by low-reorganization protein matrix in the immediate vicinity of the charge transfer zone.
From the scale of the observed effects on dielectric reorganization, the order of magnitude of possible effects corresponding to a real enzymatic reaction can be estimated. Typical values of the reorganization energy λs ≈ 60–120 kBT for charge transfer reactions are 10–20 times larger than λs ≈ 6 kBT (or 1,200 cm−1) measured for the chromophore. This is because of much stronger charge redistribution upon electron or proton transfer than upon the optical transition. The observed reduction (≈3 kBT) in the reorganization energy upon sequestering the active site inside the protein matrix corresponds to a change in λs by 30–60 kBT for the enzymatic reaction. Such a large effect is likely to play an important role in the catalytic activity of the enzyme.
Conclusions
At the active site of chymotrypsin, the protein matrix exhibits dielectric reorganization considerably weaker than that expected for a protein milieu with highly mobile polar groups. The dielectric reorganization is comparable to that in a low-polarity solvent.
Water, in contrast, exhibits an anomalously strong dielectric reorganization significantly exceeding that of solvents with comparable macroscopic dielectric properties. Its dielectric reorganization is larger than the maximum allowed within macroscopic dielectric continuum theory.
Sequestering the active site from water inside the enzyme body leads to a dramatic reduction in the dielectric response to charge transfer, an important factor in high catalytic activity. Nevertheless, the residual contribution of external water to the dielectric response at the active site is still significant. Because of the water anomaly, the role of the aqueous solvent is particularly important.
Acknowledgments
We are grateful to S. Leikin, V. A. Parsegian, and D. Rau for helpful discussion of this work, to V. A. Tikhomirov for assistance in quantum chemical calculations, and to J. Feretti and A. V. Vannikov for access to their instruments. This work was supported in part by Russian Foundation for Basic Research Grants 96-04-48374 and 99-04-48652. L.I.K. acknowledges the financial support from the National Institutes of Health of his research visit to Bethesda.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050316997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050316997
References
- 1.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 2.Krishtalik L I. Mol Biol. 1974;8:91–99. [PubMed] [Google Scholar]
- 3.Krishtalik L I. Mol Biol. 1979;13:577–581. [PubMed] [Google Scholar]
- 4.Krishtalik L I. J Theor Biol. 1980;86:757–771. doi: 10.1016/0022-5193(80)90309-4. [DOI] [PubMed] [Google Scholar]
- 5.Bailey S. Trans Faraday Soc. 1951;47:509–517. [Google Scholar]
- 6.Maricic S, Pifat G, Pravdic V. Biochim Biophys Acta. 1964;79:293–300. [PubMed] [Google Scholar]
- 7.Takashima S, Schwan H P. J Phys Chem. 1965;69:4176–4182. doi: 10.1021/j100782a019. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka A, Ishida Y. J Polym Sci Phys Ed. 1973;11:1117–1123. [Google Scholar]
- 9.Pethig R. Dielectric and Electronic Behavior of Biological Materials. New York: Wiley; 1979. [Google Scholar]
- 10.James M N G, Sielecki A R. J Mol Biol. 1983;163:299–361. doi: 10.1016/0022-2836(83)90008-6. [DOI] [PubMed] [Google Scholar]
- 11.Artymiuk P J, Blake C C F, Grace D E P, Oatley S J, Sternberg M J E. Nature (London) 1979;280:563–568. doi: 10.1038/280563a0. [DOI] [PubMed] [Google Scholar]
- 12.Gilson M K, Honig B H. Biopolymers. 1986;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura H, Sakamoto T, Wada A. Protein Eng. 1988;2:177–183. doi: 10.1093/protein/2.3.177. [DOI] [PubMed] [Google Scholar]
- 14.Karplus M, McCammon J A. CRC Crit Rev Biochem. 1991;9:293–349. doi: 10.3109/10409238109105437. [DOI] [PubMed] [Google Scholar]
- 15.King G, Lee F S, Warshel A. J Chem Phys. 1991;95:4366–4377. [Google Scholar]
- 16.Simonson T, Perahia D, Bricogne G. J Mol Biol. 1991;218:859–886. doi: 10.1016/0022-2836(91)90273-9. [DOI] [PubMed] [Google Scholar]
- 17.Simonson T, Perahia D, Brünger A T. Biophys J. 1991;59:670–690. doi: 10.1016/S0006-3495(91)82282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith P E, Brunne R M, Mark A E, van Gunsteren W F. J Phys Chem. 1993;97:2009–2014. [Google Scholar]
- 19.Simonson T, Perahia D. Proc Natl Acad Sci USA. 1995;92:1082–1086. doi: 10.1073/pnas.92.4.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marcus R A. J Chem Phys. 1963;38:1858–1862. [Google Scholar]
- 21.Hush N S. Prog Inorg Chem. 1967;8:391–443. [Google Scholar]
- 22.Mertz E L. Chem Phys Lett. 1996;262:27–32. [Google Scholar]
- 23.Mertz E L, German E D, Kuznetsov A M. Chem Phys. 1997;215:355–370. [Google Scholar]
- 24.Mertz E L, Tikhomirov V A, Krishtalik L I. J Phys Chem. 1997;101:3433–3442. [Google Scholar]
- 25.Nicholls A, Honig B. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 26.Mertz E L, Krishtalik L I. Biofizika. 1996;41:320–325. [Google Scholar]
- 27.Parker C A. Photoluminescence of Solutions. Amsterdam: Elsevier; 1968. [Google Scholar]
- 28.Lakowicz J R. Principles of Fluorescence Spectroscopy. New York: Plenum; 1983. [Google Scholar]
- 29.Lakowicz J R, Hogen D. Biochemistry. 1981;20:1366–1373. doi: 10.1021/bi00508a051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender M L, Clement G E, Kezdy F J, d'A. Heck H. J Am Chem Soc. 1964;86:3680. [Google Scholar]
- 31.Glazer A N. Proc Natl Acad Sci USA. 1965;54:171–176. doi: 10.1073/pnas.54.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernhard S A, Lee B F, Tashian Z H. J Mol Biol. 1966;18:405–420. doi: 10.1016/s0022-2836(66)80033-5. [DOI] [PubMed] [Google Scholar]
- 33.Conti E, Rivetti C, Wonacott A, Brick P. FEBS Lett. 1998;425:229–233. doi: 10.1016/s0014-5793(98)00235-x. [DOI] [PubMed] [Google Scholar]
- 34.Havsteen B H. J Biol Chem. 1967;242:769–771. [PubMed] [Google Scholar]
- 35.Kjaer A M, Ulstrup J. J Am Chem Soc. 1987;109:1934–1942. [Google Scholar]
- 36.Khurgin Iu I, Rosliakov V Ia, Kliachko-Gurvich A L, Brueva T R. Biokhimya. 1972;37:485–492. [PubMed] [Google Scholar]
- 37.Bone S. Biochim Biophys Acta. 1987;916:128–134. doi: 10.1016/0167-4838(87)90219-6. [DOI] [PubMed] [Google Scholar]
- 38.Rupley J A, Careri G. Adv Protein Chem. 1991;41:37–172. doi: 10.1016/s0065-3233(08)60197-7. [DOI] [PubMed] [Google Scholar]
- 39.Rosliakov V Ia, Khurgin Iu I. Biokhimya. 1972;37:493–497. [PubMed] [Google Scholar]
- 40.Khurgin Iu I, Medvedeva N V, Rosliakov V Ia. Biofizika. 1977;22:1010–1014. [PubMed] [Google Scholar]
- 41.Sarfare P S, Kegeles G, Kwon-Rhee S J. Biochemistry. 1966;5:1389–1393. doi: 10.1021/bi00868a036. [DOI] [PubMed] [Google Scholar]
- 42.Frauenfelder H, Petsko G A. Biophys J. 1980;32:465–483. doi: 10.1016/S0006-3495(80)84984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter J, Steigemann W, Singh T P, Bartunik H, Bode W, Huber R. Acta Crystallogr B. 1982;38:1462–1472. [Google Scholar]
- 44.MacKerell A D, Jr, Rigler R, Nilsson L, Hahn U, Saenger W. Biophys Chem. 1987;26:247–241. doi: 10.1016/0301-4622(87)80027-3. [DOI] [PubMed] [Google Scholar]
- 45.Frauenfelder H, Petsko G A, Tsernoglou D. Nature (London) 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 46.Jortner J. Biochim Biophys Acta. 1980;594:193–230. doi: 10.1016/0304-4173(80)90001-4. [DOI] [PubMed] [Google Scholar]
- 47.Kharkats Y I, Krishtalik L I. J Theor Biol. 1985;112:221–249. doi: 10.1016/s0022-5193(85)80284-8. [DOI] [PubMed] [Google Scholar]
- 48.Renger G, Hanssum B. FEBS Lett. 1992;299:28–32. doi: 10.1016/0014-5793(92)80092-u. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H X. J Am Chem Soc. 1994;116:10362–10375. [Google Scholar]
- 50.Gray H B, Winkler J R. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 51.Soriano G M, Cramer W A, Krishtalik L I. Biophys J. 1997;73:3265–3276. doi: 10.1016/S0006-3495(97)78351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chrug A K, Weiss R M, Warshel A, Takano T. J Phys Chem. 1983;87:1683–1694. [Google Scholar]
- 53.Parson W W, Chu Z T, Warshel A. Biochim Biophys Acta. 1990;1017:251–272. doi: 10.1016/0005-2728(90)90192-7. [DOI] [PubMed] [Google Scholar]
- 54.Schulten K, Tesch M. Chem Phys. 1991;158:261–270. [Google Scholar]
- 55.Zheng C, McCammon J A, Wolynes P G. Chem Phys. 1991;158:261–270. [Google Scholar]
- 56.Yadav A, Jackson R M, Holbrook J J, Warshel A. J Am Chem Soc. 1991;113:4800–4805. [Google Scholar]
- 57.Treutlein M, Schulten K, Brünger A T, Karplus M, Deisenhofer J, Michel H. Proc Natl Acad Sci USA. 1992;89:75–79. doi: 10.1073/pnas.89.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Åqvist J, Fothergill M, Warshel A. J Am Chem Soc. 1993;115:631–635. [Google Scholar]
- 59.Marchi M, Gehlen J N, Chandler D, Newton M D. J Am Chem Soc. 1993;115:4178–4190. [Google Scholar]
- 60.Parson W W, Chu Z T, Warshel A. Biophys J. 1998;74:182–191. doi: 10.1016/S0006-3495(98)77778-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pethig R. Annu Rev Phys Chem. 1992;43:177–205. doi: 10.1146/annurev.pc.43.100192.001141. [DOI] [PubMed] [Google Scholar]
- 62.Belton P S. Prog Biophys Mol Biol. 1994;61:61–79. [PubMed] [Google Scholar]
- 63.Bryant R G. Annu Rev Biophys Biomol Struct. 1996;25:29–53. doi: 10.1146/annurev.bb.25.060196.000333. [DOI] [PubMed] [Google Scholar]
- 64.Weiner S J, Kollman P A, Case D A, Singh U C, Ghio C, Alagona G, Profeta S, Jr, Weiner P. J Am Chem Soc. 1984;106:765–784. [Google Scholar]