

Abstract
Unlike in other organisms, in trypanosomes and other Kinetoplastida the larger part of glycolysis takes place in a specialized organelle, called the glycosome. At present it is impossible to remove the glycosome without changing much of the rest of the cell. It would seem impossible, therefore, to assess the metabolic consequences of this compartmentation. Therefore, we here develop a computer experimentation approach, which we call computational cell biology. A validated molecular kinetic computer replica was built of glycolysis in the parasite Trypanosoma brucei. Removing the glycosome membrane in that replica had little effect on the steady-state flux, which argues against the prevalent speculation that glycosomes serve to increase flux by concentrating the enzymes. Removal of the membrane did cause (i) the sugar phosphates to rise to unphysiologically high levels, which must have pathological effects, and (ii) a failure to recover from glucose deprivation. We explain these effects on the basis of the biochemical organization of the glycosome. We conclude (i) that the glycosome protects trypanosomes from the negative side effects of the “turbo” structure of glycolysis and (ii) that computer experimentation based on solid molecular data is a powerful tool to address questions that are not, or not yet, accessible to experimentation.
The Embden–Meyerhof pathway of glucose metabolism (glycolysis) is widespread. In most organisms the glycolytic enzymes reside in a single cellular compartment, the cytosol. An intriguing exception is trypanosomes and other organisms of the order Kinetoplastida, where the first part of glycolysis takes place in specialized organelles, called glycosomes (Fig. 1) (1). Trypanosoma brucei is the parasite that causes African sleeping disease. When living in the mammalian bloodstream, this unicellular eukaryote is completely dependent on glycolysis for its ATP production. Bloodstream-form trypanosomes contain 65 (2) to 250 (3) glycosomes, comprising 4% of the total cellular volume (3). The glycosomal membrane is impermeable to metabolites and coenzymes (4–7). Glycolytic enzymes that are highly regulated in other organisms appear to be unregulated in trypanosomes. For example, glucose 6-phosphate (Glc-6-P) strongly inhibits mammalian hexokinase (HK) I, II, and III (8) but does not affect trypanosome HK (9). The importance of regulation of yeast HK was demonstrated by a mutant that does not synthesize the yeast HK inhibitor trehalose 6-phosphate (10). When this mutant was challenged with glucose, the ATP concentration dropped, whereas Glc-6-P accumulated (11–14). It has been shown that there is a danger of unrestricted accumulation of intermediates if the design of a pathway is such that ATP is invested before its net production. The investment of ATP makes the first reactions virtually irreversible and thereby insensitive to the rest of the pathway. Therefore, a tight regulation of the first steps of such pathways is vital (15). Until now it is clear neither why trypanosomes do not need such regulation nor whether and how the compartmentation of glycolysis is advantageous to trypanosomes.
Figure 1.

A scheme of glycolysis in bloodstream-form Trypanosoma brucei. Solid lines represent reactions catalyzed by a single enzyme; dashed lines represent multiple sequential reactions. Glc-6-P, glucose 6-phosphate; Fru-1,6-BP, fructose 1,6-bisphosphate; GA-3-P, glyceraldehyde 3-phosphate; 1,3-BPGA, 1,3-bisphosphoglycerate; 3-PGA, 3-phosphoglycerate; DHAP, dihydroxyacetone phosphate; Gly-3-P, glycerol 3-phosphate.
It has been shown experimentally that correct compartmentation of glycolytic enzymes is required for viability of T. brucei (7). Glycosomes seem to be essential organelles for trypanosomes. This possibility is suggested by the observation that cell lines in which both alleles of the peroxin gene were deleted could not be obtained. Peroxin 11 is a protein involved in the biogenesis of glycosomes (16). It has been speculated that the glycosome enables trypanosomes to maintain their high glycolytic flux. Confinement of the enzymes to a small volume was proposed to overcome a postulated diffusion limitation (5, 17, 18). However, calculations based on the catalytic turnover numbers and concentrations of the glycolytic enzymes and on diffusion coefficients made a diffusion limitation highly unlikely, even if the enzymes were dispersed over the total cell volume (19, 20). The latter work has, however, not yet led to a new hypothesis explaining why compartmentation of glycolysis is advantageous. A direct comparison between the physiology of organisms with and without glycosomes is unlikely to yield such an explanation either, because these organisms are also highly different in many other respects. However, an approach that combines experimentation with computation might be fruitful. The kinetics of the glycolytic enzymes of bloodstream-form T. brucei are known and have been used to develop a detailed and validated mathematical model of its glycolysis (21, 22). We here use this computer replica of the real T. brucei glycolysis to investigate what would happen if trypanosome glycolysis were not compartmentalized.
Materials and Methods
Model Description.
Two models of trypanosome glycolysis, differing only in the presence versus absence of the glycosome, were compared. The model with the glycosome has been described previously (21). We implemented the modifications in ref. 22. A complete set of equations and parameters is available as supplementary material to this article on the PNAS web site (www.pnas.org). The model without the glycosome contains the following modifications of the original compartmentalized model:
(i) Metabolite transporters across the glycosomal membrane are absent.
(ii) There is only a single pool of ATP, ADP, and AMP, rather than distinct glycosomal and cytosolic pools.
(iii) The sum of bound phosphates is not conserved.
(iv) The same amounts of the same enzymes have been diluted into the whole cytosol.
This led to the following set of differential equations.
 |
1 |
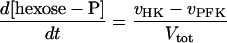 |
2 |
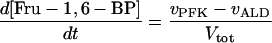 |
3 |
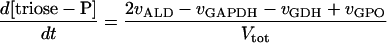 |
4 |
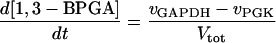 |
5 |
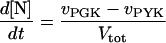 |
6 |
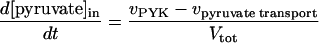 |
7 |
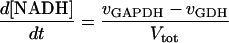 |
8 |
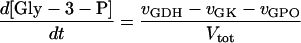 |
9 |
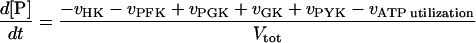 |
10 |
in which:
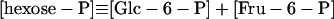 |
11 |
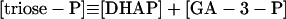 |
12 |
and
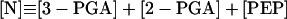 |
13 |
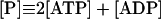 |
14 |
In these equations PFK is phosphofructokinase, ALD is aldolase, GAPDH is glyceraldehyde-3-phosphate dehydrogenase, GDH is glycerol-3-phosphate dehydrogenase, GPO is glycerol-3-phosphate oxidase, PGK is phosphoglycerate kinase, N is defined by Eq. 13, PYK is pyruvate kinase, GK is glycerol kinase, Fru-6-P is fructose 6-phosphate, Fru-1,6-BP is fructose 1,6-bisphosphate, 1,3-BPGA is 1,3-bisphosphoglycerate, 3-PGA is 3-phosphoglycerate, 2-PGA is 2-phosphoglycerate, PEP is phosphoenolpyruvate, Gly-3-P is glycerol 3-phosphate, and P is defined by Eq. 14. The enzyme rates v were expressed in nmol⋅min−1⋅(mg protein)−1, the time t in min, the metabolite concentrations in mM, and the total cell volume Vtot in μl⋅(mg protein)−1. The enzyme kinetic equations and parameters were the same in both models and have been described previously (ref. 21, with the modifications in ref. 22). The concentrations of ATP, ADP, and AMP were calculated by solving a set of three equations—i.e., Eq. 14, the conservation equation for adenine nucleotides, and the equilibrium equation of adenylate kinase (see supplementary material for details).
All simulations applied to aerobic conditions, at zero concentrations of the products, pyruvate and glycerol. The sum of the concentrations of ATP, ADP, and AMP was taken to be 4 mM, both in the cytosol in both models and in the glycosome in the model containing it. The Vmax of pyruvate export from the cells was 200 nmol⋅min−1⋅(mg cell protein)−1, as in ref. 22, where this modification should have been mentioned as well.
Software.
All simulations were performed with mlab (Civilized Software, Bethesda, MD). The time-dependent simulations were done with a Gear–Adams algorithm. Steady states were solved by a Marquardt–Levenberg algorithm.
Determination of Enzyme Concentrations in Saccharomyces cerevisiae.
S. cerevisiae strain CEN.PK122 was cultivated in batch cultures on glucose as the carbon source. Enzyme activities and glycolytic flux were measured off-line as described previously (23). Enzyme concentrations were calculated from measured enzyme activities and in vitro kinetics of purified enzymes (24–31).
Results
The Glycosome Does Not Increase the Flux by Concentrating the Enzymes.
Since it has been proposed that the high glycolytic flux in trypanosomes is due to the compartmentation of glycolysis (5), the first question we addressed was whether the glycolytic flux became higher if the enzymes converting glucose to 3-phosphoglycerate were confined to the glycosome rather than dispersed in the cytosol. On the basis of previous calculations (19, 20), diffusion was taken to be not rate-limiting. The steady-state flux was calculated as a function of the blood glucose concentration, both with and without the glycosome. Only at glucose concentrations exceeding 5 mM was the glycolytic flux slightly higher in the presence of the glycosome than in its absence (4% at 10 mM). In view of the fact that this small effect was noticed only at glucose concentrations that are unphysiologically high, and were probably not encountered by ancestral kinetoplastids, we consider it unlikely that this has provided the selective advantage for the origin of the glycosome.
An argument in favor of the hypothesis that the glycosome serves to increase the glycolytic flux has been the observation that in other organisms with a high flux, yet lacking the glycosome, the glycolytic enzymes represent a much larger part of the total cellular protein (5). To allow such a comparison, however, the flux and the amount of glycolytic enzymes should be measured under the same conditions. When this was done, it turned out that batch-grown S. cerevisiae cells have a 2-fold higher flux than does T. brucei and a less than 2-fold higher glycolytic-protein content (Table 1). This finding demonstrates that an organism that lacks the glycosome can reach a glycolytic flux per amount of glycolytic protein similar to that of T. brucei.
Table 1.
Amounts of glycolytic enzymes compared to glycolytic flux for bloodstream-form T. brucei and S. cerevisiae
| Glycolytic enzyme | % of total protein
|
|
|---|---|---|
| T. brucei | S. cerevisiae | |
| Hexokinase | 0.25 | 0.08 |
| Glucose-6-phosphate isomerase | 0.15 | 0.32 |
| Phosphofructokinase | 0.39 | 0.23 |
| Aldolase | 1.2 | 0.64 |
| Triose-phosphate isomerase | 0.04 | 0.07 |
| Glyceraldehyde-3-phosphate dehydrogenase | 0.50 | 2.5 |
| Phosphoglycerate kinase | 0.16 | 0.53 |
| Glycerol-3-phosphate dehydrogenase | 0.25 | — |
| Alcohol dehydrogenase | — | 0.59 |
| Total | 2.9 | 5.0 |
| μmol⋅min−1⋅(mg protein)−1 | ||
| Glycolytic flux | 0.15 | 0.33 |
Data for T. brucei are from refs. 18 and 22. Only the glycosomal enzymes of T. brucei were taken into account. For yeast, alcohol dehydrogenase was taken rather than glycerol-3-phosphate dehydrogenase, since it fulfills the same function (glycolytic redox balance). Glycolytic flux was defined as glucose consumption rate, which equals half the aerobic pyruvate production rate (22) in the case of T. brucei and half the anaerobic ethanol production rate in the case of S. cerevisiae. When T. brucei is compared to S. cerevisiae, the glycolytic flux is almost proportional to the amount of glycolytic protein.
The Glycosome Does Prevent Extreme Accumulation of the Hexose Phosphates.
In search for possible consequences of the compartmentation of glycolysis, the steady-state concentrations of all metabolites were calculated both in the presence and in the absence of the glycosome. Substantial differences between these two conditions were found for the substrates and products of HK and phosphofructokinase (PFK) (Fig. 2). In the glycosome the concentrations of Glc-6-P and fructose 1,6-bisphosphate (Fru-1,6-BP) remained within a narrow range, independently of the extracellular glucose concentration. When the glycosomal membrane was removed, however, Glc-6-P and Fru-1,6-BP accumulated up to 100 mM. Were this to happen in reality, the cells would probably undergo osmotic swelling and burst or suffer from phosphate depletion.
Figure 2.

In trypanosomes lacking the glycosomal membrane, hexose phosphates accumulate to extremely high levels. The steady-state intracellular concentrations of glucose (a), Glc-6-P (b), and Fru-1,6-BP (c) were calculated both for the presence (solid lines) and for the absence (dashed lines) of the glycosomal membrane. (d) Glycosomal (long-dashed line) and (solid line) cytosolic [ATP]/[ADP] ratio for if the glycosome is present and the cytosolic [ATP]/[ADP] ratio for if there is no glycosome (short-dashed line).
There were two causes of the difference between the cells with and without a glycosome. First, the sum of the concentrations of the organic phosphate that is not exchanged with inorganic phosphate is conserved within the glycosome (22). Consequently the hexose phosphates cannot accumulate above the fixed total of glycosomal organic phosphate. In the absence of the glycosomal membrane this restriction disappears, so that the hexose phosphates can accumulate. Second, the glycosomal [ATP]/[ADP] ratio behaved completely independently of the cytosolic [ATP]/[ADP] ratio (Fig. 2d). When the extracellular glucose concentration was increased, the intracellular glucose concentration followed (Fig. 2a), thereby stimulating HK. In the absence of the glycosome, HK and PFK were further activated by the increasing cytosolic [ATP]/[ADP] ratio. Because these kinases are only weakly inhibited by their products Glc-6-P and Fru-1,6-BP, respectively (9, 32, 33), these metabolites could accumulate to very high concentrations before a steady state was reached. When HK and PFK were confined to the glycosome, they were inhibited by the decreasing glycosomal [ATP]/[ADP] ratio and, consequently, a steady state was obtained at much lower concentrations of Glc-6-P and Fru-1,6-BP.
The Glycosome Keeps the Starving Cell from Consuming the ATP Required to Reinitiate Glycolysis.
The “turbo” design of glycolysis (15) is such that ATP is invested first, before net production takes place. During starvation the cell should maintain a basal ATP level (or potential to synthesize it) to be able to utilize glucose as soon as it becomes available. Trypanosomes do not store carbohydrates. How then do they deal with short periods of starvation? Within the glycosome the sum of the concentrations of phosphorylated metabolites is conserved, since the phosphate group is transferred from one compound to another, but not converted to inorganic phosphate (21, 22). The question arises whether this pool of phosphorylated metabolites can serve as the free-energy storage required to start up glycolysis after brief starvation.
To test this hypothesis computer-model cells were subjected to a low (“starvation”) glucose concentration until a steady state was reached. Suddenly, the glucose concentration was increased to a normal level (5 mM). If the initial glucose concentration was too low (0.01 mM), the uncompartmentalized cells did not recover when glucose became available (Fig. 3a; dashed line). The cells containing a glycosome rapidly increased their cytosolic ATP concentration to a normal level (Fig. 3a; solid line). At 0.01 mM glucose the cytosolic ATP concentration had dropped to zero, irrespective of the presence of the glycosome (Table 2). Consequently, if HK resided in the cytosol, it had no ATP available to phosphorylate glucose when it was taken up. Meanwhile the glycosomal ATP concentration sufficed to saturate HK (Table 2) and to reinitiate glycolysis. If the glucose concentration was not lower than 0.05 mM during the preincubation, the uncompartmentalized cells maintained a sufficiently high ATP concentration to allow 8% of the maximal HK activity (Table 2). This activity allowed glycolysis to start again, albeit hesitantly (note the initial drop in ATP) and more slowly than in cells with a glycosome (Fig. 3b)
Figure 3.

The glycosome helps trypanosomes to recover from a low blood-glucose level when returning to a normal one. Trypanosomes consuming glucose in a steady state at 0.01 mM (a) or 0.05 mM (b) extracellular glucose, were given 5 mM glucose after 60 s. The cytosolic ATP concentration was monitored, both in the model with the glycosome (solid line) and in the model without the glycosome (dashed line). (Inset) Part from 30 to 90 s enlarged.
Table 2.
At a low glucose concentration the ATP concentration can become too low to start up glycolysis in the absence of the glycosome
| Glycosome | [Glc]out, mM | Cytosolic conc., mM
|
Glycosomal conc., mM
|
HK saturation, % | ||||
|---|---|---|---|---|---|---|---|---|
| [ATP] | [ADP] | [AMP] | [ATP] | [ADP] | [AMP] | |||
| With | 0.01 | 0.00 | 0.18 | 3.82 | 4.00 | 0.00 | 0.00 | 97 |
| 0.05 | 0.08 | 0.74 | 3.18 | 3.99 | 0.01 | 0.00 | 97 | |
| Without | 0.01 | 0.00 | 0.00 | 4.00 | — | — | — | 0 |
| 0.05 | 0.06 | 0.68 | 3.25 | — | — | — | 8 | |
The steady-state adenine nucleotide concentrations have been calculated at different extracellular glucose concentrations. The percentage saturation of HK with ATP was calculated as 100%·([ATP]/Km,ATPHK)/(1 + [ATP]/Km,ATPHK + [ADP]/Km,ADPHK), in which Km,ATPHK was 0.116 mM and Km,ADPHK was 0.126 mM (9). In the absence of a glycosome the cytosolic concentrations of ATP and ADP were used, and if HK was localized in the glycosome, the glycosomal concentrations of these compounds were used.
In general, the time needed for the transition from one steady state to another depends not only on the activities of the enzymes but also on the size of the metabolite pools they have to fill (34). Glycosomes constitute only 4% of the total cellular volume (3). Consequently, the metabolite pools to be filled are smaller if part of glycolysis is localized in the glycosome. We wondered whether this volume effect also contributed to the more rapid transition time of the compartmentalized glycolysis (Fig. 3b). This was not the case. When the glycosomal volume was varied between 50% and 200% of its normal volume (at constant concentrations of the conserved sums, either at constant total volume or at constant cytosolic volume) no effect was observed on the dynamics of the cytosolic ATP concentration during the transition from 0.05 to 5 mM glucose.
Discussion
We have investigated the consequences of compartmentation of glycolysis by comparing two models of T. brucei glycolysis: one with a glycosome and one in which the glycosomal enzymes and cofactors had been diluted into the whole cytosol. Here we shall discuss whether the differences between these two conditions provide clues concerning a possible function of the glycosome.
Our calculations do not support the prevailing hypothesis that the glycosome serves to maintain a high glycolytic flux (5, 17, 18): cells with and without compartmentation consumed glucose almost equally rapidly over a wide range of blood glucose concentrations. In a simple, linear metabolic pathway compartmentation should not affect the steady-state flux at all, unless the transport of metabolites across the compartment membrane controls the flux. In trypanosome glycolysis, however, the situation is more complicated, because the cytosolic kinases interact with an adenine-nucleotide pool that differs from the one with which the glycosomal kinases interact. The glycosomal ATP concentration is determined by the balance between its production by phosphoglycerate kinase and glycerol kinase and its consumption by HK and PFK. The cytosolic ATP concentration is balanced by pyruvate kinase and the utilization of ATP in free-energy-requiring processes. If the glycosomal membrane is removed, there is only a single ATP concentration, which is balanced by all above processes together. This caused the slight difference between the glycolytic flux in the presence versus absence of the glycosomal membrane, but it did not lead to any substantial difference. In fact, although T. brucei metabolizes glucose very rapidly as compared with several mammalian cell types, its glycolysis is not particularly fast compared with some other organisms that lack the glycosome (20). Moreover, most other Kinetoplastida, which all carry glycosomes, do not have such a high flux.
One conspicuous difference between trypanosomes and other eukaryotes is the lack of regulation of HK and PFK in the former. Unrestricted accumulation of intermediates tends to occur especially if enzymes in the beginning of a pathway are kept far from equilibrium by the continuous investment of ATP, as is the case in many catabolic pathways, including glycolysis. Therefore, tight regulation of such enzymes is necessary (15). Indeed, the products of the unregulated trypanosome HK and PFK accumulated up to 100 mM when the glycosomal membrane was removed in our calculations. This “phenotype” was reminiscent of the S. cerevisiae mutant that is unable to synthesize the HK inhibitor trehalose 6-phosphate (11). When the first part of glycolysis took place in glycosomes, the hexose phosphates did not accumulate. This finding is consistent with experiments: the highest estimated glycosomal concentrations based on experimental data are 4 mM Glc-6-P, 2 mM fructose 6-phosphate, and 2 mM Fru-1,6-BP (35). The explanation is that the kinase activities inside the glycosome were restrained by the decreasing glycosomal [ATP]/[ADP] ratio in the organelle. When the glycosomal membrane was removed the kinases were stimulated by the increasing cytosolic [ATP]/[ADP] ratio. Thus the compartmentation of the adenine-nucleotide pool in trypanosomes can substitute for the tight regulation of the kinases as found in yeast and mammals (15, 36).
From phylogenetic trees it has been concluded that the glycosomal enzymes diverged from their homologues in other eukaryotes early in evolution (37, 38). While the core structure of the glycolytic enzymes and their catalytic mechanisms have been conserved extremely well during evolution, their regulation differs substantially among the different phylogenetic groups (39). Possibly, when Kinetoplastida acquired the glycosome, the glycolytic enzymes of the latter were less tightly regulated than are the present-day enzymes of other eukaryotes. Compartmentation of glycolysis should have given these cells an advantage at that time (40). After acquiring the glycosome, Kinetoplastida may have lost all, by then redundant, regulation, thereby turning the compartmentation of glycolysis into an irreversible event.
The investment of ATP in the initial steps of a pathway may lead not only to overactive first steps but also to a situation in which the pathway cannot get started because of a depletion of ATP. This indeed occurred in the model where glycolysis was not compartmentalized. In contrast, if the first enzymes were localized in the glycosome, the conserved glycosomal pool of phosphorylated metabolites ensured a sufficiently high local ATP concentration to utilize the available glucose after a short period of starvation. For T. brucei this may be relevant, because it does not contain any storage carbohydrates to guarantee a basal ATP production when an exogenous carbon source is lacking. In this study it was assumed that the pool of phosphorylated compounds was completely conserved, but in reality this pool may be emptied slowly by hydrolysis of, for example, glycosomal ATP or 1,3-bisphosphoglycerate. If so, this pool can serve only transiently as an ATP-equivalent storage device. Consistently, T. brucei does survive starvation, but only for 10 min at 37°C (41). Leishmania and Crithidia species, other members of the order Kinetoplastida, survive several hours of carbon starvation (42, 43), perhaps because they contain the storage compound polymannose (mannan).
It is difficult to infer from the above results which selective advantage a specialized glycolytic organelle offered to the ancestral kinetoplastids. Two possible advantages that glycosomes may have for present-day bloodstream-form trypanosomes were identified in this paper: (i) they serve as an alternative to a tight regulation of HK and PFK and (ii) they ensure a sufficiently high ATP level to initiate glycolysis after a brief period of starvation. Of course, the fact that the glycosome provided the observed advantages does not prove that these are the functions of the glycosome. But then, the same could be said for any corresponding evidence obtained by removing the glycosomal membrane experimentally.
The result that, in the absence of the glycosome, glycolysis should be hazardous to the cell is in itself surprising. It is because of the autocatalytic nature of the glycolytic pathway: two ATP molecules are invested to make the beginning of the pathway irreversible, but four molecules of ATP are recovered at the end. In principle, this can lead to an autocatalytic acceleration of the pathway and an “explosion,” or at least a gross derangement such as would have been caused by 200 mM hexose phosphates. On the other side of the spectrum of possibilities, an autocatalytic pathway like this can be trapped in a “stalling” or “death state” when the concentration of ATP becomes too low to get the pathway started (44). Many catabolic (i.e., free-energy-harvesting) pathways are autocatalytic in the above sense. In the case of glycolysis it has been shown that this is optimal for flux (45). Accordingly, organisms need gadgets that prevent the stalling and explosion side effects of the optimal pathway structure. In this paper it was demonstrated that one effective gadget is the glycosome, by its nature of setting an upper and a lower limit to the total concentration of organic phosphates. Alternatives include an extensive regulation of HK plus the availability of storage compounds, which appears to occur in yeast (15).
We reached these conclusions by a strategy that to our knowledge is new to cell biology—i.e., by computer experimentation on a model of the real system. Of course, computer modeling as such is not new to cell biology. For instance, the feasibility of cell-cycle models has been checked by mathematical modeling (46). And microtubule dynamics have been simulated with computer models (47). However, these models were neither precise nor validated as they addressed the feasibility of a hypothesis. We emphasize that the computer experimentation performed here is a special type of computer modeling. For most computer modeling one does not need precise knowledge of all parameters of the system under study. Some or even all parameters can be fitted; the question then addressed is whether one can obtain a computer model that simulates the observed behavior. By contrast, computer experimentation is meant to substitute for true experimentation. Accordingly it has to be based on precise kinetic knowledge of the molecular components of the system, rather than on fitted parameter values. Only when this condition is met can the calculated result be trusted to be realistic. In our example here, the required manipulation was the removal of the glycosomal membrane without otherwise affecting the cell. At present this is not a feasible physical experiment, but it was possible in the computer replica of reality. The latter could be constructed only because all relevant kinetic parameters had been determined experimentally. In contrast to the usual computer modeling, the computer experimentation discovered hitherto unknown physiological behavior. Additional discoveries may pertain to effects of drugs, to the optimal compartmentation of metabolism, and to interference with trypanosome metabolism in the presence of glycerol.
This use of computer experimentation is not new to science as a whole. In fact, it is the α and ω of computational physics. And in biology the method is known in the area of molecular dynamics and in the calculation of structures on the basis of NMR or x-ray data. However, to cell biology it is new, and it may indeed be a bit unexpected that one can calculate the consequences of a membrane from enzyme kinetic data. New as it may be, the method may prove increasingly useful in the era of functional genome analysis.
Supplementary Material
Acknowledgments
We thank Drs. F. R. Opperdoes and J. T. Pronk for very stimulating discussions and suggestions. This study was supported by the Netherlands Organization for Scientific Research (NWO) and the Netherlands Association of Biotechnology Research Schools (ABON).
Abbreviations
- HK
hexokinase
- PFK
phosphofructokinase
- Glc-6-P
glucose 6-phosphate
- Fru-1,6-BP
fructose 1,6-bisphosphate
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030539197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030539197
References
- 1.Opperdoes F R, Borst P. FEBS Lett. 1977;80:360–364. doi: 10.1016/0014-5793(77)80476-6. [DOI] [PubMed] [Google Scholar]
- 2.Tetley L, Vickerman K. J Microsc. 1991;162:83–90. doi: 10.1111/j.1365-2818.1991.tb03118.x. [DOI] [PubMed] [Google Scholar]
- 3.Opperdoes F R, Baudhuin P, Coppens I, De Roe C, Edwards S W, Weijers P J, Misset O. J Cell Biol. 1984;98:1178–1184. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visser N, Opperdoes F R, Borst P. Eur J Biochem. 1981;118:521–526. doi: 10.1111/j.1432-1033.1981.tb05550.x. [DOI] [PubMed] [Google Scholar]
- 5.Opperdoes F R. Annu Rev Microbiol. 1987;41:127–151. doi: 10.1146/annurev.mi.41.100187.001015. [DOI] [PubMed] [Google Scholar]
- 6.Clayton C E, Michels P A M. Parasitol Today. 1996;12:465–471. doi: 10.1016/s0169-4758(96)10073-9. [DOI] [PubMed] [Google Scholar]
- 7.Blattner J, Helfert S, Michels P A M, Clayton C. Proc Natl Acad Sci USA. 1998;95:11596–11600. doi: 10.1073/pnas.95.20.11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colowick S P. In: The Enzymes. Boyer P D, editor. Vol. 9. New York: Academic; 1973. pp. 1–48. [Google Scholar]
- 9.Nwagwu M, Opperdoes F R. Acta Trop. 1982;39:61–72. [PubMed] [Google Scholar]
- 10.Blazquez M A, Lagunas R, Gancedo C, Gancedo J M. FEBS Lett. 1993;329:51–54. doi: 10.1016/0014-5793(93)80191-v. [DOI] [PubMed] [Google Scholar]
- 11.Thevelein J M, Hohman S. Trends Biochem Sci. 1995;20:3–10. doi: 10.1016/s0968-0004(00)88938-0. [DOI] [PubMed] [Google Scholar]
- 12.Luyten K, De Koning W, Tesseur I, Ruiz M C, Ramos J, Cobbaert P, Thevelein J M, Hohmann S. Eur J Biochem. 1993;217:701–713. doi: 10.1111/j.1432-1033.1993.tb18296.x. [DOI] [PubMed] [Google Scholar]
- 13.Hohmann S, Bell W, Neves M J, Valckx D, Thevelein J M. Mol Microbiol. 1996;20:981–991. doi: 10.1111/j.1365-2958.1996.tb02539.x. [DOI] [PubMed] [Google Scholar]
- 14.Hohmann S, Neves M J, De Koning W, Alijo R, Ramos J, Thevelein J M. Curr Genet. 1993;23:281–289. doi: 10.1007/BF00310888. [DOI] [PubMed] [Google Scholar]
- 15.Teusink B, Walsh M C, Van Dam K, Westerhoff H V. Trends Biochem Sci. 1998;23:162–169. doi: 10.1016/s0968-0004(98)01205-5. [DOI] [PubMed] [Google Scholar]
- 16.Lorenz P, Maier A G, Baumgart E, Erdmann R, Clayton C. EMBO J. 1998;17:3542–3555. doi: 10.1093/emboj/17.13.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aman, R. A., Kenyon, G. L. & Wang, C. C. (1985) 260, 6966–6973. [PubMed]
- 18.Misset O, Bos O J M, Opperdoes F R. Eur J Biochem. 1986;157:441–453. doi: 10.1111/j.1432-1033.1986.tb09687.x. [DOI] [PubMed] [Google Scholar]
- 19.Westerhoff H V, Welch G R. Curr Top Cell Regul. 1992;33:361–390. doi: 10.1016/b978-0-12-152833-1.50026-5. [DOI] [PubMed] [Google Scholar]
- 20.Bakker B M, Westerhoff H V, Michels P A M. J Bioenerg Biomembr. 1995;27:513–525. doi: 10.1007/BF02110191. [DOI] [PubMed] [Google Scholar]
- 21.Bakker B M, Michels P A M, Opperdoes F R, Westerhoff H V. J Biol Chem. 1997;272:3207–3215. doi: 10.1074/jbc.272.6.3207. [DOI] [PubMed] [Google Scholar]
- 22.Bakker B M, Michels P A M, Opperdoes F R, Westerhoff H V. J Biol Chem. 1999;274:14551–14559. doi: 10.1074/jbc.274.21.14551. [DOI] [PubMed] [Google Scholar]
- 23.Van Hoek P, Van Dijken J P, Pronk J T. Appl Environ Microbiol. 1998;64:4226–4233. doi: 10.1128/aem.64.11.4226-4233.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnard E A. Methods Enzymol. 1975;42:6–20. doi: 10.1016/0076-6879(75)42085-7. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa Y, Noltmann E A. J Biol Chem. 1965;240:L1877–L1881. [PubMed] [Google Scholar]
- 26.Stellwagen E, Wilgus H. Methods Enzymol. 1975;42:78–85. doi: 10.1016/0076-6879(75)42097-3. [DOI] [PubMed] [Google Scholar]
- 27.Rutter W J, Hunsley J R, Groves W E, Calder J, Rajkumar T V, Woodfin B M. Methods Enzymol. 1966;4:479–486. [Google Scholar]
- 28.Nickbarg E B, Knowles J R. Biochemistry. 1988;27:5939–5947. doi: 10.1021/bi00416a018. [DOI] [PubMed] [Google Scholar]
- 29.Byers L D. Methods Enzymol. 1982;89:326–335. doi: 10.1016/s0076-6879(82)89059-9. [DOI] [PubMed] [Google Scholar]
- 30.Scopes R K, Griffiths-Smith K, Miller D G. Anal Biochem. 1981;118:284–285. doi: 10.1016/0003-2697(81)90583-2. [DOI] [PubMed] [Google Scholar]
- 31.Scopes R K. Methods Enzymol. 1975;42:134–138. doi: 10.1016/0076-6879(75)42106-1. [DOI] [PubMed] [Google Scholar]
- 32.Cronin C N, Tipton K F. Biochem J. 1985;227:113–124. doi: 10.1042/bj2270113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cronin C N, Tipton K F. Biochem J. 1987;245:13–18. doi: 10.1042/bj2450013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Easterby J S. Biochem J. 1981;199:155–161. doi: 10.1042/bj1990155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misset O, Opperdoes F R. Eur J Biochem. 1984;144:475–483. doi: 10.1111/j.1432-1033.1984.tb08490.x. [DOI] [PubMed] [Google Scholar]
- 36.Iynedjian P B. Trends Biochem Sci. 1998;23:467–468. doi: 10.1016/s0968-0004(98)01317-6. [DOI] [PubMed] [Google Scholar]
- 37.Michels P A M, Opperdoes F R, Hannaert V, Wiemer E A C, Allert S, Chevalier N. Belg J Bot. 1992;125:164–173. [Google Scholar]
- 38.Michels P A M, Opperdoes F R. Parasitol Today. 1991;7:105–109. doi: 10.1016/0169-4758(91)90167-m. [DOI] [PubMed] [Google Scholar]
- 39.Fothergill-Gilmore L A, Michels P A M. Prog Biophys Mol Biol. 1993;59:105–235. doi: 10.1016/0079-6107(93)90001-z. [DOI] [PubMed] [Google Scholar]
- 40.Michels P A M, Hannaert V. J Bioenerg Biomembr. 1994;26:213–219. doi: 10.1007/BF00763070. [DOI] [PubMed] [Google Scholar]
- 41.Seyfang A, Duszenko M. Eur J Biochem. 1991;202:191–196. doi: 10.1111/j.1432-1033.1991.tb16362.x. [DOI] [PubMed] [Google Scholar]
- 42.Keegan F P, Blum J J. Mol Biochem Parasitol. 1992;53:193–200. doi: 10.1016/0166-6851(92)90021-b. [DOI] [PubMed] [Google Scholar]
- 43.Blum J J. J Bioenerg Biomembr. 1994;26:147–155. doi: 10.1007/BF00763063. [DOI] [PubMed] [Google Scholar]
- 44.Heinrich R, Rapoport S M, Rapoport T A. Prog Biophys Mol Biol. 1977;32:1–82. [PubMed] [Google Scholar]
- 45.Heinrich R, Montero F, Klipp E, Waddell T G, Melendez-Hevia E. Eur J Biochem. 1997;243:191–201. doi: 10.1111/j.1432-1033.1997.0191a.x. [DOI] [PubMed] [Google Scholar]
- 46.Novak B, Tyson J J. Proc Natl Acad Sci USA. 1997;94:9147–9152. doi: 10.1073/pnas.94.17.9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pantaloni D, Hill T L, Carlier M F, Korn E D. Proc Natl Acad Sci USA. 1985;82:7207–7211. doi: 10.1073/pnas.82.21.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.