

Abstract
With fs time, speed, and angular resolution of the elementary steps in electron transfer reactions, we report direct observation of reversible and dissociative processes for dative bonding involving covalent and ionic characters. For bimolecular reactions of various donors and acceptors we find strong correlation between the structure and the dynamics. The dynamics from the transition state to final products involve two elementary processes, with different reaction times, speed, and angular distributions. For example, for the R2S⋅I2 (R = C2H5) system, it is shown that after charge separation, the reversible electron transfer occurs in less than 150 fs (fastest trajectory) and is followed by the rupture of the I—I bond with the release of the first I-atom in 510 fs. However, the second process of the remaining and trapped I-atom takes 1.15 ps with its speed (500 m/s) being much smaller than the first one (1,030 m/s). The S—I—I average angle is 130°. These findings, on this and the other systems reported here, elucidate the mechanism and address some concepts of nonconcertedness, caging, and restricted energy redistribution.
In the description of the chemical bond, the contribution of covalent and ionic characters is essential to energetics and dynamics and hence to the physical and chemical properties. Such a concept was evident in early theories of chemical bonding by Lewis (1), Pauling (2), Mulliken (3), and others. In many classes of reactions, where the covalent and ionic potential energy surfaces become close in energy, the description of the dative bond takes into account the two possible forms, and the wave function becomes (4):
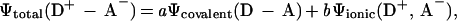 |
where the covalent and ionic structures are distinct for the donor (D)–acceptor (A) complex and the coefficients a and b depend on the ability of D to give an electron (ionization potential) and A to accept the electron (electron affinity).
The nature of these charge-transfer complexes was described by Mulliken in 1952 (3) after Benesi and Hildebrand in 1949 (5) observed a new absorption band in a solution of benzene and iodine dissolved in n-heptane. Since then there have been numerous studies in the liquid, gas, and solid phases (6, 7). Ultrafast studies in solutions have revealed the dissociation and caging dynamics on different time scales (8–11). Under the isolated, binary condition of D and A there have been few studies. Only recently have the dynamics been studied (12, 13) for this famous benzene-iodine system. Studies of charge-transfer and related processes have provided a microscopic picture in clusters (14) under controlled solvation (15–17), in a precursor-determined geometry (18–20), and in matrices (21).
For isolated electron transfer (ET) reactions, the dynamics are unique in that the nature of the bond changes with time. With fs time resolution it is possible to prepare the system in a covalent (or ionic) structure and observe the evolution to the ionic (covalent) structure and perhaps the reversible ET to the covalent (ionic) structure. In the process, chemical bonds may form or break and the dynamics will elucidate such chemical changes following ET. Such processes, relevant in many electrochemical and surface studies (22–25), are important to the mechanism, which involves the following central questions: What is the time scale for ET and its possible reversible pathway? Are the subsequent chemical events concerted or nonconcerted with ET? What is the geometrical structure in the transition-state region? What is the nature of the reaction trajectory, coherent or incoherent?
In a series of ET reactions, we report direct observation of the temporal evolution and the resolution of the speed and angular distributions of the reaction at different times, using fs kinetic-energy resolved time-of-flight (KETOF) mass spectrometry, as described in refs. 26 and 27. The isolated, bimolecular reactions were studied in a molecular beam, varying the donors and acceptors to control the energetics, i.e., the location of the charge-transfer state. Specifically, we studied diethyl sulfide, dioxane, acetone, and benzene as donors, and iodine and iodine monochloride as acceptors. In Fig. 1, we present the concept of the experiment, illustrated for the reaction of diethyl sulfide (D) and iodine (A). The fs pulse prepares the ionic structure at t = 0 from the neutral, ground-state structure. We then follow the dynamics after charge separation by monitoring the product (I) as a function of time, kinetic energy (speed), and orientation.
Figure 1.

A generic representation of the potential energy of ET reactions with the ionic and covalent changes along the reaction coordinate. At zero time, the initial wave packet is launched at the ionic configuration from the ground-state structure, R2S⋅⋅⋅I2. The four snapshots show the reaction at t < 0, after the ET in the transition-state region (t = t‡), and after reaching the final (t = tf) products along either the neutral, dissociative channel or the ionic path. Note that the reaction coordinate shown here is a path through a multidimensional potential energy surface, the contours of which are depicted in Fig. 5.
The results are striking in demonstrating that, although ET occurs at early times from D to A, the electron reversibly goes back to the donor, leaving the acceptor with enough energy to break the I—I bond. In other words, the initial ionic configuration converts following the covalent pathway. Furthermore, we observe two dynamical processes for this neutral and dissociative reaction as the system proceeds from the ionic to the covalent potential. The reaction times of the two processes are very different: the first I-atom is liberated in 510 fs, whereas the second one is trapped coherently in the force field of the “substrate” for 800 fs and completely departs in 1.15 ps. The average angle of S—I—I (see Fig. 1) is 130°. In the series studied here, we found that the time scales of ET and reactions are critical for the effective dimensionality of the nuclear motion and the degree of concertedness.
METHODOLOGY
Experimental.
The experimental apparatus is described in detail elsewhere (26, 27). All experiments were performed in a two-chamber molecular-beam apparatus integrated with a tunable fs laser system. Two fs laser pulses, delayed in time, were spatially overlapped in the extraction field region of a two-stage linear TOF mass spectrometry. The axes of the molecular and laser beams and the TOF are mutually orthogonal. The pump laser pulse was centered at 277 nm and the probe at ≈304 nm for a 2+1 resonance-enhanced multiphoton ionization process of free I-atoms.
A gas mixture containing diethyl sulfide (0.4 Torr vapor), iodine (1 Torr), and helium (800 Torr) was expanded through a pulsed valve. All other systems were handled similarly. The TOF mass spectrum was obtained for all systems reported here and only under the 1:1 complex condition were the studies made. The I-atom transient signal was enhanced by a factor of ≈14 when the diethyl sulfide was added, indicating a direct charge-transfer excitation. The enhancement is in accord with the much stronger absorption of the R2S⋅I2 complex than those of I2 and R2S (28) and excludes the contributions from the locally excited R2S*⋅I2 and R2S⋅I2* reactions.
Velocity and Angular Resolution.
The translation energy release because of the bond breakage causes a spread in the KETOF profile around the central time T0 (in TOF). As the time spread (ΔT = T − T0) is a linear function of the three-dimension velocity projection onto the TOF axis (z), the KETOF profile is transformed into the velocity (vz) distribution by the equation vz = −(ΔTqE)/m, where m and q are the mass and charge of the ionized I-atom and E is the extraction electric field strength. As detailed in ref. 27, this velocity distribution is sensitive to three parameters of dynamics: the speed distribution g(v) determined by the energetics of the bond rupture; the anisotropy β determined on the fs time scale, where no rotation is present, by the direction (θ) of the recoil velocity relative to the initial alignment (transition dipole); β = 2P2(cosθ), where P2 is the second Legendre polynomial; and the angle χ of the pump-pulse linear polarization with respect to the TOF axis. By recording the KETOF distribution at the magic angle (χ = 54.7°), the speed distribution is directly derived and by altering the angle χ, the anisotropy and the corresponding angle (θ) are obtained. Typically, we resolve the I-atom KETOF profile at χ = 54.7° to directly obtain the speed distributions. We then observe the flux of these atoms for different polarizations of the fs pump pulse relative to the TOF axis.
RESULTS AND DISCUSSION
Time and Velocity Correlations.
The fs-resolved KETOF distributions at the magic angle (χ = 54.7°) are shown in Fig. 2A, and the KETOF distributions for three different pump polarizations (χ) at the reaction time of 7.1 ps are given in Fig. 2B. Two distinct velocity distributions, fast and slow, are clearly present and show a dramatic difference in the temporal behavior. At early times, only the fast I-atoms appear and the distribution reaches a plateau value after 1.75 ps. However, the slow ones show up only after ≈900 fs and gradually build up until 7 ps. Because of the dramatic difference in the temporal behavior, the two distributions are not resulting from the parallel reaction pathways, e.g., for the covalent channel I/I* + R2S⋅I/R2S⋅I* involving the two spin-orbit states, I and I* (separated by 7,600 cm−1). Instead, the two distributions are from two different dynamical processes of the reaction.
Figure 2.

(A) The fs-resolved KETOF distributions at the magic angle (χ = 54.7°). Note the different temporal behavior for the two velocity distributions. (B) The KETOF distributions of the I-atoms at the three different angles (χ) at 7.1-ps reaction time. The open circles are the experimental data and the solid lines are the results of the theoretical simulation. The simulation procedure (27) only uses the first half (vz ≤ 0) of the KETOF distribution; because of symmetry, the second half (vz ≥ 0) of the data shown here are the replica of the first half.
In Fig. 3, the two velocity distributions are revealed in the speed distributions, derived from the magic-angle (χ = 54.7°) data (Fig. 2A) (27). The two distributions are well separated at v = 800 m/s (ET = 3,420 cm−1). The high-speed component is centered at ≈1,030 m/s (5,700 cm−1) and extends to 1,300 m/s (10,000 cm−1). This fast component is unambiguously assigned to the first I-atom released through the covalent channel, based on the following energetics consideration.
Figure 3.

The fs-resolved speed distributions of the I-atoms obtained from the χ = 54.7° magic-angle data (Fig. 2A). The derived final anisotropy distribution and the corresponding angle between the recoil direction (the bond orientation) and the initial fs alignment are shown at the top. Note the shift of the slow component with time and the lack thereof in the fast component, as indicated by the dashed lines.
The maximum charge-transfer absorption of the R2S⋅I2 complex in the gas phase is at 290 nm (28). Recent ab initio calculations give an equilibrium distance of 3.32 Å between S and I and a binding energy of −6.2 kcal/mol (29). For a complete ET, the R2S+I− equilibrium distance is derived (12) to be 3.7 Å. To account for a fraction ET, we take the range of 2.7 to 3.7 Å, predicting that the lower limit of the zero-point energy of R2S+I− is ≈2.1 eV above its ground state. Therefore, the upper limit of the available energy for the ionic channel is ≈0.6 eV at 277-nm excitation, and the maximum translational energy of the released I-atoms, based on the kinematics, is ≈3,000 cm−1, much smaller than our observed 10,000 cm−1 for the first I-atom. The presence of such covalent channel indicates that reversible ET is responsible for the very high speed acquired by the fragments, much higher than that of the ionic channel.
The slow distribution is centered at a final speed of v = 500 m/s (1,335 cm−1) but terminates at 300 m/s (480 cm−1). Furthermore, the peak shifts (Fig. 3) from the maximum of v = 620 m/s at early times to v = 500 m/s after 7 ps, indicating the degree of inelastic energy transfer from the atomic motion to the donor molecule. The slow I-atom release is also from the covalent channel, and any contribution from the ionic one must be negligible. This conclusion is supported by four observations: the difference in anisotropy (recoil direction) and difference in coherent caging time from the fast I-atom distribution (shown below), the shift of the entire distribution with time (shown above), and the fact that the I-atom distribution observed here did not extend to zero speed, typically observed in many harpoon reactions (30, 31). Accordingly, this slow component is also from the covalent channel, released after collisions with R2S in a one-molecule caging process.
The temporal behavior of the two processes is also very distinct. The gating of the fast or slow component gives the transient shown in Fig. 4A with the temporal behavior not only different for the fast and slow I-atoms but also from that observed collecting all I-atoms. The total I-atom signal gives the time constant τ = 850 ± 50 fs for the overall reaction time. The fast component, however, gives a rise of 510 fs with a ≈150-fs coherent delay from the time zero, whereas the slow component shows a ≈800-fs coherent shift and a rise time of 1.15 ps.
Figure 4.

Fs-gating of different speed distributions. Total iodine atom transients (○) and the gated transients (●) of the fast and slow components are shown for the different systems studied. The solid lines are the theoretical results using a coherent delay and exponential rise describing the time evolution (see text). The iodine molecule was used as the electron acceptor with the donor structures shown, except for E where the electron acceptor was iodine monochloride and the donor was benzene. Note the dramatic difference in reaction times and coherent delays.
The 510-fs reaction time of the first I-atom represents the prompt rupture of the bond in the D-A system, i.e., the transition-state lifetime of R2S+⋅⋅⋅− I⋅⋅⋅I‡. The coherent shift of 150 fs is the shortest time for a typical trajectory moving from the initial well-localized configuration to the product of R2S⋅I+I. For the second process, the 1.15-ps reaction time represents the average lifetime of the R2S⋅I collision complex, and the large coherent shift (800 fs) indicates that the second I-atom was trapped in the force field of the donor (R2S). As many as 43% of the caged iodine atoms are not liberated from the collision complex of R2S⋅I, as our observed branching ratio of the fast I-atoms to the slow ones is 100:57 (Fig. 4A, at long times).
Anisotropy and the Structure.
The initial structure was elucidated by measurements of the vectorial correlation between the direction of recoil and the initial fs alignment, which is particularly the case for the fs time resolution as there is no rotation on this time scale. The final recoil anisotropy β(v) and the corresponding angle (θ) are shown at the top of Fig. 3. The β(v) distribution for the two components is quite different. The average β for the fast component is ≈0.4 and the corresponding angle is 47°. However, the slow I-atoms have a higher, nearly constant βs ≈0.95, indicating that all released slow I-atoms have nearly the same direction and the resulting angle is 144°.
The recent ab initio calculation (29) predicts a tetrahedral structure with a nearly linear configuration of S—I—I. The iodine molecule mainly interacts with the lone-pair electrons of the sp3 orbital of the sulfide atom. According to Mulliken’s resonance theory (3), the overall transition-dipole moment in the strong complex (≥6 kcal/mol) is dominated by the dipole moment of the charge-transfer state, and the direction points, in this case, from the sulfide atom to the center of mass of the iodine molecule. Because the fastest I-atoms (v ≈ 1,300 m/s) are liberated on the fs time scale, it is reasonable to consider the complex structure as the initial one. From β of the fastest, first I-atom release we obtain the angle to be 36°, which describes the initial structure because of the fs reaction time. Note that after ET, essentially all the energy is in the I—I bond (from energy conservation) and there are no other significant vibrations to alter the structure on this fs time scale. Accordingly, we derive an experimental value of 130° for the angle of S—I—I, i.e., a nonlinear structure (see Fig. 1 at t < 0). For the fast I-atoms we observed (Fig. 3) the following correlation between β and v: the lower the speed, the smaller the anisotropy and the larger the corresponding angle, indicating the structural changes with energy release.
Reaction Dynamics and the Mechanism.
The mechanism involving the two elementary processes is now elucidated. After the vertical fs excitation at t = 0, the I2 anion is produced in high vibrational levels, around the dissociation limit (32). The entire complex begins the nuclear motion toward the hybrid region of covalent and ionic potentials (Figs. 1 and 5). However, in the region where the covalent repulsive (R2S⋅I⋅⋅⋅I) and the Coulomb ionic (R2S+I−⋅⋅⋅I and R2S+⋅⋅⋅−I—I) potentials are near crossing, the electron reversibly goes to R2S, leaving the I—I bond on the dissociative covalent potential. The observed 150-fs coherent delay time of the fast I-atoms is simply the direct bond-breaking time of I2 on the repulsive surface. The 510-fs rise time indicates that the complex executes several vibrations before all trajectories finally hop from the ionic to the covalent surface by a spontaneous (reversible) ET from the acceptor to the donor. Our observed broad energy distribution of the first I-atom (≈7.4 kcal/mol, full width at half-maximum) is basically caused by the distribution of multiple ionic-covalent crossings (Figs. 1 and 5) and the three possible exit dissociation asymptotes, I/I* + R2S⋅I/R2S⋅I*.
Figure 5.

The potential energy contour map for two of the nuclear coordinates involved, the distance between the donor and acceptor and between the two atoms of the acceptor, both for the ionic pathway. The potential is drawn for ET transfer of 75%, which determines the equilibrium position of the well; see text. The seam (the result of the repulsive I⋅⋅⋅I separation and ionic potentials) also is shown. A trajectory, from molecular dynamics calculations, is shown, together with the three snapshots of the structure at t = 0 (initial wave packet; solid oval), at the seam (thick curved line), and in the final ionic product channel. Note that the trajectory has multiple crossings to the covalent potential and, in this case, this route is the dominant pathway of ET reactions.
After the direct I—I bond rupture, the second caging process is controlled by the collisions between the remaining I-atom and R2S. The time scale for this process is determined by the relative collision energy and the interaction force. The caged iodine, whose initial configuration is dictated by the geometry after the I—I bond breakage, is launched at a repulsive, anisotropic potential surface (Fig. 5) between R2S and I (azimuthal and radial directions). From measurement of the recoil angle of the first I-atom we obtained ≈145°-120° for the angle between the trapped I-atom velocity and the direction of the initial fs alignment. The initial distribution of collision energies of 4.0–11.9 kcal/mol (average ≈6.7 kcal/mol), relative to the binding energy (29, 33) of R2S⋅I (≈6 kcal/mol), supports the finding that the R2S⋅I complexes are trapped in the potential well. The execution of several bending motions before liberating the I-atom results in the 800-fs coherent delay time, which represents the minimum time for any escaped trajectory to move from the initial well-localized configuration to the final product of R2S+I, including any resonance motion during the energy redistribution. A similar time behavior also was observed for the CH3I⋅I complex, with a 1.4-ps coherent delay time (34). From the kinematics of the collision, the fraction of the second I-atom energy transferred in the caging process is ≈35%. This inelastic energy transfer is evident in the temporal shift of the slow-speed component with time, as discussed above (Fig. 3).
Similar reactions were studied by changing the donor to acetone, dioxane, and benzene, and the acceptor to iodine monochloride. The I-atom transients are shown in Fig. 4 B–D. Two distinct speed distributions and two different time scales were observed, generalizing the above picture to all systems studied. We note the following correlation: for the second process of I-atom liberation, the larger the binding energy, the longer the coherent delay time and the smaller the branching ratio of this process to the first one. For the reaction of benzene and iodine monochloride (ICl, acceptor), the temporal behavior (Fig. 4E) is unique. Only one slow I-atom distribution was observed, indicating that the iodine (not chlorine) faces the benzene in the complex and that it experiences an inelastic collision after a reversible ET. This picture is consistent with the structure derived from the recent ab initio calculation (35). The findings again support the above-described reaction mechanism.
Conclusion.
The time, speed, and orientation resolution of dynamics of ET reactions is a powerful approach for dissecting the different elementary processes and elucidating the mechanism. The studies reported here emphasize several concepts. First, the reversibility of ET, which leads to a covalent pathway, is an important mechanism and could be general in the so-called harpoon reaction mechanism. Second, the separation of time scales for the two elementary processes and for ET indicates the nonconcertedness between ET and the chemical bond breakage, a point of debate in many areas of studies. Moreover, the appearance of coherent reaction trajectories is evidence of the localized nuclear wave packet motion. Third, the dynamics of energy dissipation to the donor (“substrate”) critically depends on the time scales of ET and bond breakage. On the ultrashort time scale, this energy dissipation is not dominant. In the case reported here, ≈15% of the total energy dissipates as internal energy of the substrate, R2S. Thus only few reactive coordinates can be considered because there is not enough time to reach a complete energy redistribution. This concept is important in studies of surface and surface-aligned photochemical reactions (36–38).
The interplay between the structure and the ensuing dynamics is rationalized with the help of LUMO-HOMO interactions. As depicted in Fig. 5, the initial structure, corresponding to the wave packet at zero time, is ionic with the transferred electron being in the σ* LUMO of the I—I bond. When the trajectory of the motion reaches the seam and an electron goes back to the donor, the iodine system is in an equivalent structure to that of an nσ* repulsive potential. Note that the reverse process of ET must involve a different electron otherwise the system will go back to the ground state. Thus, the reason for the dominance of trajectories on the covalent potential and the reverse of ET now can be understood. In Fig. 5, a typical calculated molecular dynamics trajectory shows that the trapping in the Coulomb potential (ionic channel) takes much longer time than the rupture on the repulsive surface (covalent channel), and hence it is the time scale of the nuclear motion that shifts the dominance toward the covalent bond breakage following ET (unpublished work).
Acknowledgments
We thank Dr. Thorsten M. Bernhardt for his help and discussion. This work is supported by a grant from the National Science Foundation, the Air Force Office of Scientific Research, and the Office of Naval Research.
ABBREVIATIONS
- ET
electron transfer
- KETOF
kinetic-energy resolved time of flight
References
- 1.Lewis G N. Valence and the Structure of Atoms and Molecules. New York: Chemical Catalog; 1923. [Google Scholar]
- 2.Pauling L. The Nature of the Chemical Bond. New York: Cornell Univ. Press; 1939. [Google Scholar]
- 3.Mulliken R S. J Am Chem Soc. 1952;74:811–824. [Google Scholar]
- 4.Mulliken R S, Person W B. Molecular Complexes: A Lecture and Reprint Volume. New York: Wiley; 1969. [Google Scholar]
- 5.Benesi H A, Hildebrand J H. J Am Chem Soc. 1949;71:2703–2707. [Google Scholar]
- 6.Yarwood J. Spectroscopy and Structure of Molecular Complexes. New York: Plenum; 1973. [Google Scholar]
- 7.Foster R. Molecular Complexes. London: Elek Science; 1973. [Google Scholar]
- 8.Langhoff C A, Gnädig K, Eisenthal K B. Chem Phys Lett. 1980;46:117–121. [Google Scholar]
- 9.Hilinski E F, Rentzepis P M. J Am Chem Soc. 1985;107:5907–5910. [Google Scholar]
- 10.Lenderink E, Duppen K, Wiersma D A. Chem Phys Lett. 1993;211:503–510. [Google Scholar]
- 11.Pullen S, Walker L A, II, Sension R J. J Chem Phys. 1995;103:7877–7886. [Google Scholar]
- 12.Cheng P Y, Zhong D, Zewail A H. J Chem Phys. 1996;105:6216–6248. [Google Scholar]
- 13.DeBoer G, Burnett J W, Fujimoto A, Young M A. J Phys Chem. 1996;100:14882–14891. [Google Scholar]
- 14.Castleman A W, Jr, Bowen K H., Jr J Phys Chem. 1996;100:12911–12944. [Google Scholar]
- 15.Lineberger W C, Nadal M, Nandi S, Wenthold P, Kim J B, Andersen L H, Ozaki Y, Boo D W. In: Femtochemistry and Femtobiology: Ultrafast Reaction Dynamics at Atomic-Scale Resolution. Sundström V, editor. Singapore: Imperial College Press; 1997. pp. 423–435. [Google Scholar]
- 16.Greenblatt B J, Zanni M T, Neumark D M. Science. 1997;276:1675–1678. [Google Scholar]
- 17.Ayotte P, Bailey C G, Weddle G H, Johnson M A. J Phys Chem A. 1998;102:3067–3071. [Google Scholar]
- 18.Jouvet C, Boivineau M, Duval M C, Soep B. J Phys Chem. 1987;91:5416–5422. [Google Scholar]
- 19.Nelson T O, Setser D W, Qin J. J Phys Chem. 1993;97:2585–2595. [Google Scholar]
- 20.Chang X Y, Ehlich R, Hudson A J, Polanyi J C, Wang J-X. J Chem Phys. 1997;106:3988–4001. [Google Scholar]
- 21.Zadoyan R, Apkarian V A. Chem Phys Lett. 1993;206:475–482. [Google Scholar]
- 22.Eberson L. Electron Transfer Reactions in Organic Chemistry. Berlin: Springer; 1987. [Google Scholar]
- 23.Savéant J M. Adv Phys Org Chem. 1990;26:1–131. [Google Scholar]
- 24.Marcus R A. Angew Chem Int Ed Engl. 1993;32:1111–1121. [Google Scholar]
- 25.Fox, M. A., ed. (1992) Chem. Rev.92, 365–490.
- 26.Zewail A H. Femtochemistry: Ultrafast Dynamics of the Chemical Bond. Singapore: World Scientific; 1994. , and references therein. [Google Scholar]
- 27.Zhong D, Zewail A H. J Phys Chem A. 1998;102:4031–4058. [Google Scholar]
- 28.Tamres M, Goodenow J M. J Phys Chem. 1967;71:1982–1989. [Google Scholar]
- 29.Ammal S S C, Ananthavel S P, Chandrasekhar J, Venuvanalingam P, Hegde M S. Chem Phys Lett. 1996;248:153–157. [Google Scholar]
- 30.Tamagake K, Kolts J H, Setser D W. J Chem Phys. 1979;71:1264–1275. [Google Scholar]
- 31.Donovan R J, Greenhill P, MaDonald M A, Yencha A J, Hartree W S, Johnson K, Jouvet C, Kvaran A, Simon J P. J Chem Soc Faraday Discuss. 1987;84:221–238. [Google Scholar]
- 32.Chen E C M, Wentworth W E. J Phys Chem. 1985;89:4099–4105. [Google Scholar]
- 33.Su J T, Zewail A H. J Phys Chem A. 1998;102:4082–4099. [Google Scholar]
- 34.Zhong D, Cheng P Y, Zewail A H. J Chem Phys. 1996;105:7864–7867. [Google Scholar]
- 35.Ammal S S C, Ananthavel S P, Venuvanalingam P, Hegde M S. J Phys Chem A. 1998;102:532–536. [Google Scholar]
- 36.Garrett S J, Heyd D V, Polanyi J C. J Chem Phys. 1997;106:7847–7855. [Google Scholar]
- 37.Ho W. Surf Sci. 1994;299/300:996–1007. [Google Scholar]
- 38.Hellberg L, Strömquist J, Kasemo B, Lundqvist B I. Phys Rev Lett. 1995;74:4742–4745. doi: 10.1103/PhysRevLett.74.4742. [DOI] [PubMed] [Google Scholar]