

Abstract
The mammalian retromer complex consists of SNX1, SNX2, Vps26, Vps29, and Vps35, and retrieves lysosomal enzyme receptors from endosomes to the trans-Golgi network. The structure of human Vps26A at 2.1Å resolution reveals two curvedβ -sandwich domains connected by a polar core and a flexible linker. Vps26 has an unexpected structural relationship to arrestins. The Vps35-binding site on Vps26 maps to a mobile loop spanning residues 235–246, near the tip of the C-terminal domain. The loop is phylogenetically conserved and provides a mechanism for Vps26 integration into the complex that leaves the rest of the structure free for engagements with membranes and for conformational changes. Hydrophobic residues and a Gly in this loop are required for integration into the retromer complex and endosomal localization of human Vps26, and for the function of yeast Vps26 in carboxypeptidase Y sorting.
The biosynthetic sorting of acid hydrolase precursors from the trans-Golgi network (TGN) to the endosomal-lysosomal system is central to the biogenesis of lysosomes in metazoans. In mammals, this sorting is directed by binding of mannose 6-phosphate groups on the hydrolases to two transmembrane receptors known as the cation-dependent and cation-independent mannose 6-phosphate receptors (CD-MPR and CI-MPR, respectively) 1. At the TGN, the hydrolase-receptor complexes are packaged into carrier vesicles by several coat and adaptor proteins, including clathrin, AP-1 (adaptor protein 1) and GGA (Golgi-localized, gamma ear-containing, ADP ribosylation factor-binding) proteins 1,2. These carrier vesicles bud from the TGN and fuse with endosomes. Within the endosome, the acid pH in the lumen triggers the release of the hydrolases from their receptors. The hydrolases are carried with the fluid phase to lysosomes, while the receptors return to the TGN to be reutilized in further rounds of sorting. Several proteins and complexes, including AP-1 3,4, TIP47 (tail-interacting protein of 47 kDa) 5 and PACS-1 (phosphofurin acidic cluster sorting protein) 6 have been implicated in the transport of MPRs from endosomes to the TGN.
A similar process has been described in yeast cells for the sorting of acid hydrolases to the vacuole, which is the fungal equivalent of the mammalian lysosome. Carboxypeptidase Y (CPY) is sorted by the Vps10 transmembrane receptor. Genetic screens in yeast have identified more than 60 Vps (vacuolar protein sorting) gene products 7, which are involved in the transport of CPY to the vacuole. Five yeast Vps proteins, Vps5, Vps17, Vps26, Vps29 and Vps35, form a complex named “retromer” that is required for Vps10 sorting from endosomes to the late-Golgi 8. Vps5 and Vps17 belong to the sorting nexin family and form a distinct subcomplex 9–11. Vps5 and Vps17 contain Phox homology (PX) and BAR (Bin/amphiphysin/Rvs) domains. PX domains target proteins to endosomal membranes by binding the lipid phosphatidylinositol 3-phosphate 12–16, and BAR domains are banana-shaped modules that target to curved vesicular or tubular membranes through their concave face 17. Vps35 and Vps29 form a stable cargo recognition subcomplex, with Vps35 directly binding to the Vps10 cytosolic tail 18.
A homologous retromer complex consisting of five subunits termed SNX1, SNX2, Vps26, Vps29 and Vps35 has been described in humans 8. Depletion of some of these subunits in vivo by RNA interference (RNAi) impairs retrieval of the CI-MPR and results in its missorting to lysosomes, where the receptor is degraded 19–21. The membrane targeting of mammalian retromer seems to be mediated, at least in part, by the sorting nexins, SNX1 and SNX2 20,22. Vps35 directly interacts with the CI-MPR cytoplasmic tail suggesting that it constitutes a cargo-recognition subunit of the complex 19. Recently, the structures of mouse and human Vps29 were determined, revealing that Vps29 has an inactive form of a divalent metal-containing phosphoesterase fold 23,24. Vps29 uses two different exposed hydrophobic patches to bind to Vps35 and sorting nexins, respectively 24.
The function of the Vps26 subunit has been the least clear of any of the five subunits. There are no homologous proteins of known structure or function, and few clues as to its role within the complex. Vps26 is required for embryonic development in mice25, and is downregulated in Alzheimer’s disease26, highlighting its importance in mammalian physiology. Two isoforms of Vps26, Vps26A and Vps26B are encoded by the mouse genome, and both are capable of interacting with other retromer subunits 27. To further understand the assembly of the retromer and the function of Vps26 in retrograde trafficking, we solved the structure of human Vps26A (herein referred to as Vps26) at 2.1Å by x-ray crystallography. The structure reveals that Vps26 is a close structural relative of the arrestins, an extensively characterized family of proteins involved in receptor internalization at the plasma membrane 28. Vps26 shares not only the same overall fold as the arrestins, but an unusual polar core as well. Using a combination of structure-based mutagenesis and yeast two-hybrid assays, we show that Vps26 interacts directly with Vps35 through a loop near the distal tip of the C-terminal domain. This interaction is essential for the assembly of Vps26 into the retromer complex and its recruitment to endosomes in mammalian cells, and for function of the retromer complex in vacuolar sorting of CPY in yeast. The combination of structural homology, the known properties of the arrestins, and the mapping of the Vps35 binding site onto the structure allows us to advance a working model for the function of this subunit.
RESULTS
Structure determination of Vps26
The full-length 327-residue human Vps26 protein crystallizes in space group P212121. The crystal structure was determined by using multiwavelength anomalous dispersion from both SeMet-substituted protein crystals and Pt-soaked derivative crystals, together with isomorphous replacement using native and Pt derivative crystals (Supp. Fig. 1). Vps26 behaves as a monomer in solution during purification and packs as one molecule per asymmetric unit in the crystal. Within the crystal, tubes that run the length of the crystal are formed where concavities in each Vps26 monomer are juxtaposed to each other (Supp. Fig. 2). The refined model contains residues 6 to 299 with a gap between 239 and 244 due to missing electron density. Residue 161 on the linker between two domains also shows poor density. The final model was refined to 2.1Å with Rfree=28% and R-factor=23%.
Molecular architecture of Vps26
The Vps26 molecule consists of two domains; each of them folds into a deeply curved β- sandwich ( Fig. 1a). Each sandwich is formed by a four-stranded antiparallel β-sheet packed against an antiparallelβ-sheet of three or four strands in the N-terminal domain (N domain) and C-terminal domain (C domain), respectively. The N domain contains residues 6–148, and the C domain includes residues 164–299. There is a 15-residue loop, from 149 to 163 (L10), that connects the two domains. This loop is relatively mobile, with poor electron density for several residues centered on Asn161. The N domain and the C domain are related by an intramolecular pseudo-two-fold rotation axis, with the r.m.s.d. of 1.5 Å when superimposing the N domain onto C domain using 59 Cα atoms from core β-strands. The surface of the inner β-sheet of each domain is polar. Residues at the outer layer of the β-sandwich from the C domain display high B factors (above 70 Å2 on average) with residues 239 to 244 missing from the electron density map, indicating that this region is more mobile than the rest of the protein.
Figure 1. Structure of Vps26A.
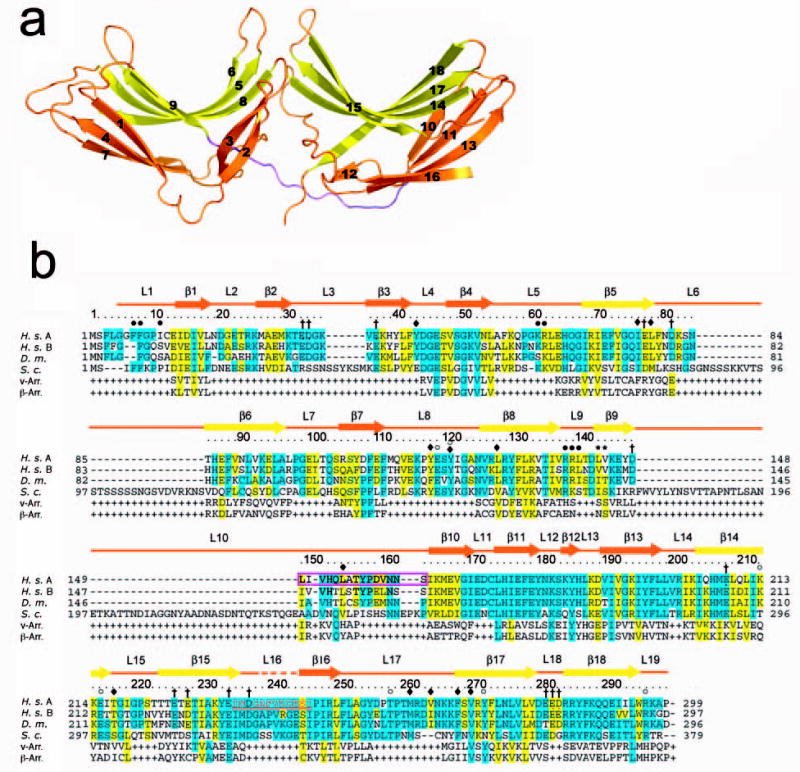
(a) Ribbon model of Vps26. Inner β-strands are colored in yellow and outer β-strands are colored in orange. The interdomain linker is colored in magenta. All structural figures were generated with Pymol (W. Delano, www.pymol.org). (b) Structure-based sequence alignment of Vps26 orthologs and arrestins (visual arrestin PDB: 1CF1 and β-arrestin 1 PDB: 1G4M). Residues involved in Vps35 binding are colored in red; polar core residues are labeled by open circles ○; hydrophobic residues in the interdomain contact are labeled by filled diamonds♦; acidic patch residues are labeled by daggers †; basic patch residues are labeled by filled circles•; yeast dominant negative mutants are labeled with an asterisk *; and the N-C domain linker is boxed in magenta.
Vps26 is electrically polarized. Its surface contains a prominent basic patch and two acidic patches ( Fig. 2a). The residues in this patch are conserved from yeast to human ( Fig. 1b). The second acidic patch straddles the interface between two domains and is only partially conserved in yeast. An electropositive patch at the far end of the N domain consists of well-conserved residues Lys61, Arg62, Arg138, and Arg139 ( Fig. 2a,b). Furthermore, several Phe residues protrude into solution immediately adjoining the basic patch ( Fig. 2c). This combination of exposed hydrophobic and basic residues is a hallmark of membrane-binding structures 29, and suggested to us that Vps26 could interact with membranes via this motif. We have tested binding to a range of liposomes containing PI3P and other acidic lipids using surface plasmon resonance, however, and have been unable to observe any noticeable interaction as compared to a series of well-characterized control proteins (data not shown). While the Vps26 N domain could bind to membranes too weakly to localize to membranes on its own, it could be a secondary interaction site in the context of the larger complex as assembled on the membrane.
Figure 2. Surface properties of Vps26.
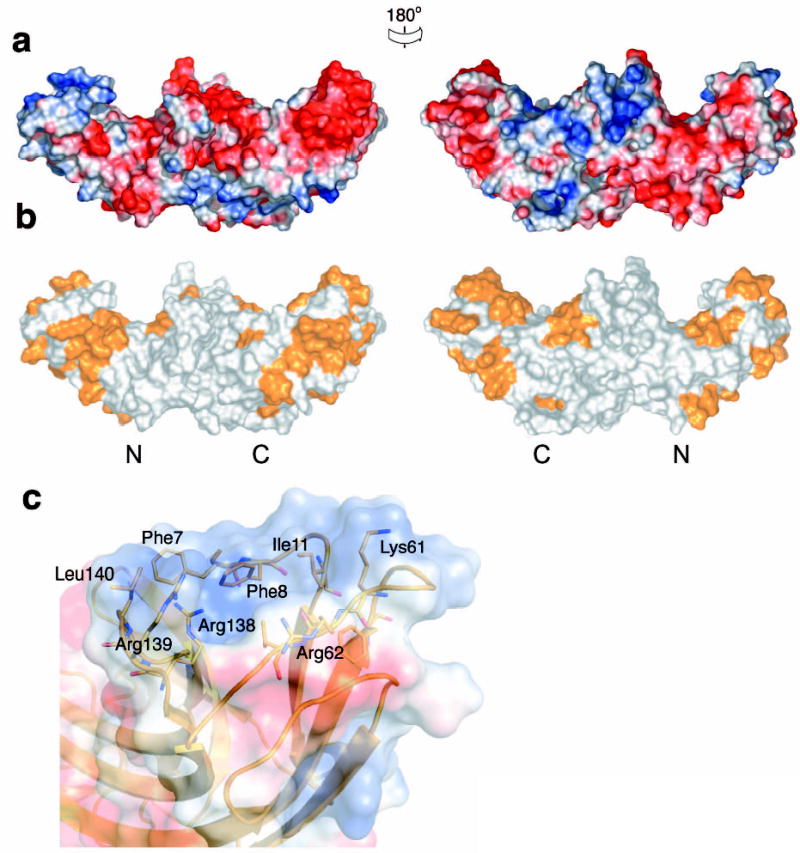
(a) Electrostatic potential. Surfaces are colored with saturating blue and red at ± 5kT/e. (b) Residues altered in site-directed mutagenesis ( Table 1) are colored in orange. (c) Potential membrane binding site showing in surface representation and colored with saturating blue and red at ± 5kT/e. Hydrophobic residues and positively charged residues are shown in orange balls and sticks.
The interface between the N domain and the C domain buries 1964 Å2 of solvent accessible surface area ( Fig. 3). The interface buries 700Å2 polar surface and 1264Å2 non-polar surface. The hydrophobic interaction is formed by residues from the body of N domain, the body of C domain and the linker loop. Twelve hydrophobic residues participate in this interaction ( Fig. 3a). An extensive buried polar core is formed between the two domains. The polar core includes N domain residues Glu119 and Tyr121, and C domain residues Lys213, Glu215, Thr258, Tyr272, and Arg296 ( Fig. 1b, 3b, c). The most extensive buried hydrogen bond network occurs between the side-chains of Tyr121, Lys213 and Glu215, and main chain amino groups from Ala124 and Asn125 ( Fig. 3b). The second network is formed by a buried salt bridge between Glu119 and Arg296 ( Fig. 3c). With the exception of Lys213, residues involved in forming these interactions are highly conserved in Vps26 proteins, suggesting that they have a critical role in protein function ( Fig. 1b).
Figure 3. Polar core of Vps26.
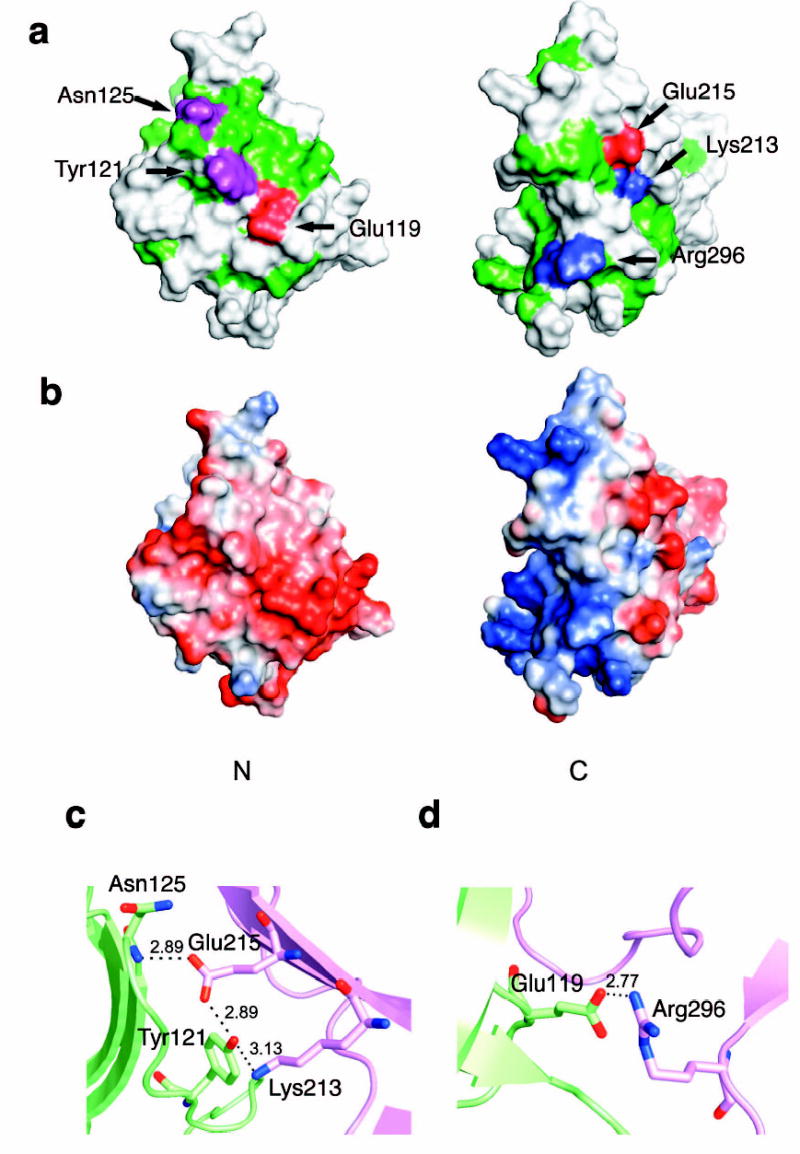
(a) Polarity of the interface between two domains. Hydrophobic residues are colored in green, polar residues are colored in white. Acidic residues buried in the polar core are colored in red, uncharged residues involved in hydrogen bonding are colored magenta, and basic residues are colored in blue. (b) Electrostatic potential at the interface is colored with saturating blue and red at ± 5kT/e. (c) Residues contributing to the first cluster of polar interactions are shown in balls and sticks. Hydrogen bonds distances are in Å. N domain residues are colored in lime and C domain residues are colored in violet. (d) Residues contributing to the second cluster of polar interactions are shown in balls and sticks.
An unexpected structural homology to the arrestins
The fold of Vps26 closely resembles that of the arrestin family members despite the absence of detectable sequence homology ( Fig. 4), on the basis of a search of the protein data bank 30. Superimposing Vps26 with visual arrestin (v-arrestin) 31 using Cα of corresponding β-strands results in the r.m.s.d. of 1.4 Å between N domains (63 Cα atoms), and 1.5 Å between C domains (94 Cα atoms), respectively. One of the defining features of arrestin family proteins is the existence of a polar core embedded between the two domains. Vps26 shares this feature in the general sense that the domain interface contains clusters of buried polar interactions ( Fig. 4a, b). The overall extents of the polar cores are similar, although the identities of individual amino acids are not conserved ( Fig. 4c, d).
Figure 4. Structural similarities between Vps26 and β-arrestin.
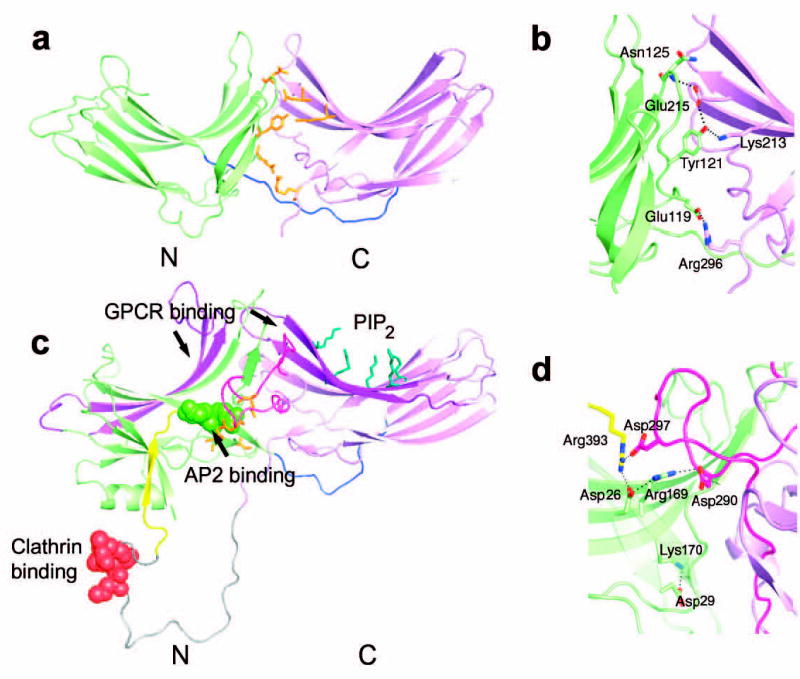
(a) Vps26 is shown in a ribbon model with the N domain colored in lime and the C domain in violet. The interdomain linker is colored blue. Polar core residues are colored in orange. (b) β -arrestin1 is shown in a ribbon model in the same color scheme as (a). The disordered region at the C tail is modeled with in an arbitrary conformation and colored in grey with the clathrin binding site shown in red spheres. The AP-2 binding site is shown in green spheres. The GPCR binding sites in the N and C domain cups are colored in purple. The PIP2 binding residues are shown in blue balls and sticks. The C domain lariat and the C terminal tail are colored in pink and yellow respectively. (c) Polar core residues in Vps26. (d) Polar core residues in β-arrestin1.
Mapping the binding site for Vps35 on Vps26
Vps26 interacts directly with Vps35 32,33. To identify the binding site for Vps35 on Vps26 we constructed 30 mutants of Vps26 carrying substitutions of surface residues (Table 1; Fig. 5; constructs #1–30). These mutations included non-conservative substitutions of single residues or clusters of 2–4 residues, and in one case the replacement of a Gly-Gly linker sequence for a loop comprising residues 238–246. The design of this latter mutation enabled deletion of the loop while still allowing the linkage of the adjoining sequences without conformational distortion of the protein. The choice of these mutation sites was based on several criteria: conservation, exposed hydrophobic residues, concave shape, correspondence to functional sites in arrestin, and unusually strong positive or negative electrostatic potential, all of which are likely interactions sites. The interaction of these Vps26 mutants with Vps35 was tested using a yeast two-hybrid system in which Vps26 variants and Vps35 were expressed as fusions with the transcription activation domain (Gal4AD) and DNA-binding domain (Gal4BD) of the Gal4 transcription factor, respectively. We observed that the mutation I235D M236D (#16) or deletion of the 238–246 loop (#18; Δ238–246 GG), abrogated interaction with Vps35, whereas the other 28 mutations had no detectable effect ( Fig. 5a). Immunoblot analysis showed that the binding-defective mutants were expressed at levels that were similar to those of wild-type Vps26 and other Vps26 mutants ( Fig. 5b). Strikingly, both of these mutations map to the same region of Vps26, the distal tip of the C domain ( Fig. 5d). The 28 other single and multiple mutations tested represent coverage of a total of 69 surface residues, including all of the likely interaction sites. The lack of any effect on the Vps35 interaction rules out these regions as candidate binding sites. We conclude that Vps35 binds to a single isolated site on Vps26 located at one tip of the molecule.
Table 1.
Summary of Vps26 mutant phenotypes
| Mutant Number | Mutations(human/yeast equivalents) | Vps35 binding in two- hybrid assay a | Assembly with Vps35, Vps29 in vivo b | Endosomal localization in cells | Rescue of CPY processing in VPS26Δ yeast d | Rescue of CPY secretion in VPS26Δ yeast e |
|---|---|---|---|---|---|---|
| – | WT | ++ | + | + | + | + |
| 1 | L431A V144D K145D | ++ | ||||
| 2 | E13A D15A | ++ | ||||
| 3 | K133D T135D V137D | ++ | ||||
| 4 | R69A E71A | ++ | + | + | ||
| 5 | L97D | ++ | ||||
| 6 | R127E R129E F131E | ++ | ||||
| 7 | F79D | ++ | ||||
| 8 | R271E F273E | ++ | ||||
| 9 | K214E I216E | ++ | ||||
| 10 | T225D T227D | ++ | ||||
| 11 | I230D Y233D | ++ | ||||
| 12 | L250D F251D | ++ | ||||
| 13 | F197D L198D | ++ | ||||
| 14 | V200D I204D | ++ | ||||
| 15 | I246D | ++ | ||||
| 16 | I235D M236D/I318D M319D | – | – | – | – | – |
| 17 | R284E R285E F287E | ++ | ||||
| 18 | Δ238–246 GG/Δ321–328 GG | – | – | – | – | – |
| 19 | V190D V192D K194D | ++ | ||||
| 20 | E177A E179A K182D | ++ | ||||
| 21 | E165D K167A | ++ | ||||
| 22 | E171A D172A | ++ | ||||
| 23 | E281A E182A D183A R284A | ++ | ||||
| 24 | S49A K51D N53A | ++ | ||||
| 25 | V137D R139D L140D | ++ | ||||
| 26 | T102D Q103D R105D | ++ | ||||
| 27 | D108A E110A M112D | ++ | ||||
| 28 | V53A A55D K57D | ++ | ||||
| 29 | K61A R62D E64D | ++ | ||||
| 30 | E234A D237A | ++ | ||||
| 31 | I235D | + | ||||
| 32 | M236D | + | ||||
| 33 | G238P/G321P | ± | – | – | – | – |
| 34 | 238–246 polyS/321–328 polyS | ± | – | – | – | – |
| 35 | Y121F/Y150F | + | + | |||
| 36 | N125R/N155D | + | + | |||
| 37 | E215R/E298R | + | + | |||
| 38 | R296E/R377E | + | + |
Yeast two-hybrid assays were performed as in Fig. 5; ++, normal growth; +, reduced growth in the presence of 10mM 3-AT; ±, lack of growth in the presence of 10mM 3-AT, -, no growth.
Co-precipitation of Vps35 and Vps29 with wild-type or mutant Vps26-myc expressed by transfection in HeLa cells was performed as in Fig. 6; +, normal assembly; -, reduced or no assembly.
Co-localization (+) or lack thereof (−) of wild-type or mutant Vps26-myc with SNX1 on endosomes in HeLa cells analyzed as in Fig. 6. For each condition, 50–75 cells from three independent experiments were examined.
CPY processing and secretion were analyzed as in Fig. 7; for both assays, +, normal function; –, reduced function.
Figure 5. Identification of the Vps35 binding site on Vps26.
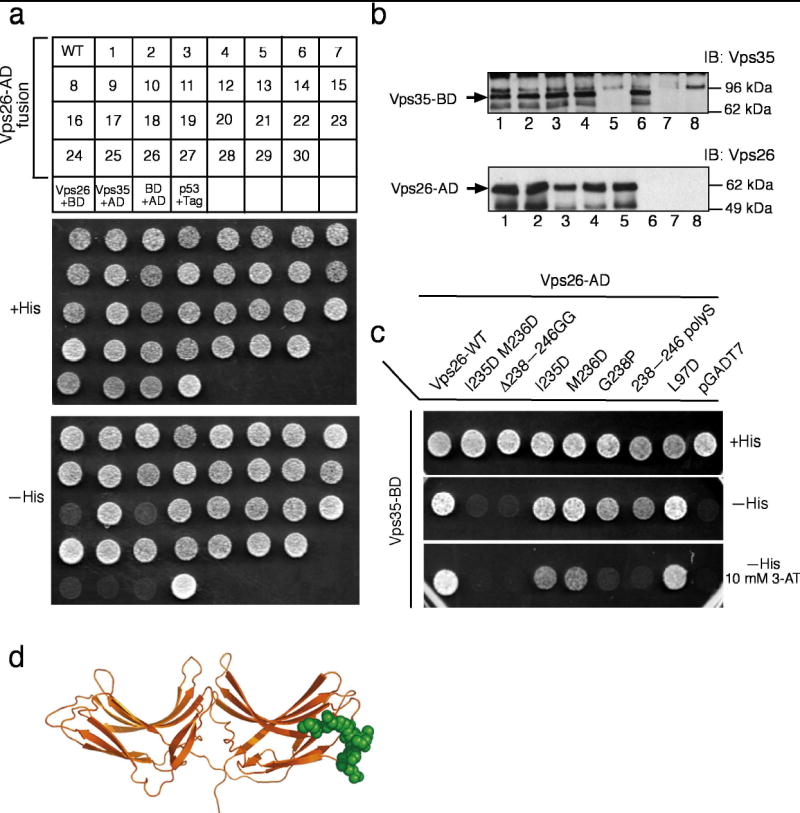
(a) The interaction of Vps35 fused to Gal4BD with wild-type or mutant Vps26 constructs (numbered from 1 to 30 in Table 1) fused to Gal4AD was analyzed using the yeast two-hybrid system. The ability to grow in the absence of histidine (-His) is indicative of interactions. A summary of the results from three independent experiments is presented in Table 1 and a representative experiment is shown in the figure. Negative and positive controls for interactions described in Materials and Methods are also shown. Notice that the I235D M236D (#16) and Δ238–246 GG(#18)Vps26 transformants fail to grow on –His plates. (b) Whole cell extracts of the co-transformed cells from the yeast two-hybrid assay expressing Vps35 together with wild-type (WT) (lane 1), I235D M236D (#16, lane 2), Δ238–246 GG(#18)lane 3) and L250D F251D (#12, lane 4) Vps26 constructs, Vps35 and pGADT7 (lane 6), pGBKT7 and wild-type Vps26 (lane 5), pGBKT7 and pGADT7 (lane 7), or p53 and SV40 large T-antigen (Tag) (lane 8) were subjected to SDS-PAGE and immunoblotting using polyclonal antibodies to Vps35 (top panel) and Vps26 (bottom panel) (c) Additional Vps26 mutants were tested for interaction with Vps26 using the yeast two-hybrid system. A summary of the results is presented in Table 2 and a representative experiment is shown in the figure. Notice that the 238–246 polyS and G238P mutants of Vps26 fail to support growth on –His plates in the presence of the competitive inhibitor of the His3 protein, 3-aminotriazole (3-AT). (c) Residues involved in binding Vps26 are highlighted in a space-filling model. Residues that are disordered in the Vps26 structure were modeled in a sterically allowed but otherwise arbitrary conformation. In panel (a) BD: pGBKY7 and AD: pGADT7.
To map the determinants of Vps26 binding to Vps35 in more detail, several additional mutations in the loop region were generated (Table 1; Fig. 5c; constructs #31–34). The histidine biosynthesis inhibitor 3-AT was used in one set of assays in order to achieve higher stringency and resolve weaker mutational effects. The entire mobile loop was replaced by a poly-Ser linker in one construction, 238–246 polyS. This mutant construct showed interaction with Vps35 in the absence of 3-AT, but not in its presence ( Fig. 5c). Similarly, substitution of loop residue Gly238 by proline (G238P) resulted in detection of interaction with Vps35 in the absence, but not the presence, of 3-AT ( Fig. 5c). A construct consisting of only loop residues 232–247 failed to interact with Vps35 in the two-hybrid assay (data not shown). We conclude that the mobile loop and adjacent residues are the primary binding site for Vps35. The loop is not autonomous, however, and it must be presented in the context of the Vps26 fold.
To determine whether the same mutations affected the assembly of Vps26 with Vps35 in human cells, we transfected HeLa cells with mammalian expression plasmids encoding myc-tagged wild-type or mutant Vps26 constructs ( Fig. 6). These constructs were then isolated by immunoprecipitation with an antibody to the myc epitope and the immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with antibodies to Vps35 and Vps29. We found that whereas the wild-type and R69A E71A Vps26 constructs co-precipitated endogenous Vps35 and Vps29, the I235D M236D, Δ238–246 GG, 238–246 polyS and G238P Vps26 constructs did not ( Fig. 6a). This was despite similar levels of expression of all recombinant myc-tagged constructs in the transfected cells ( Fig. 6a). Therefore, mutations in the loop and adjacent regions not only prevent interaction of Vps26 with Vps35 but also block the incorporation of Vps26 into the endogenous Vps26:Vps29:Vps35 subcomplex. This is consistent with the notion that Vps26 only interacts with retromer in the context of the Vps26:Vps29:Vps35 subcomplex, and defines the mechanism of Vps26 integration into the retromer.
Figure 6. The binding site for Vps35 is required for Vps26 integration into the retromer complex in vivo.

(a) Lysates from HeLa cells that were either not transfected (lanes 1 and 8, NT) or transfected with cDNAs encoding wild-type (lanes 2 and 9), IM-235,236-DD (lanes 3 and 10), Δ238–246 GG (lanes 4 and 11), 238–246 polyS (lanes 5 and 12), G238P (lanes 6 and 13) and R69A E71A (lanes 7 and 14) forms of myc-tagged Vps26 were subjected to immunoprecipitation (IP) using a mouse monoclonal antibody to the myc epitope The lysates (1% of the total, lanes 1–7) and immunoprecipitates (lanes 8–14) were subsequently analyzed by SDS-PAGE and immunoblotting (IB) with antibodies to the myc epitope (top panel), Vps35 (middle panel) or Vps29 (bottom panel). (b) The intracellular localization of Vps26-myc and the Vps26-myc mutants indicated in the figure (left column shows Alexa 488 green channel), as well as the colocalization of these constructs with SNX1 (middle column shows Alexa 546, red channel), was examined in fixed/permeabilized cells by indirect immunofluorescence and confocal microscopy. The right-hand column shows merged images; yellow indicates co-localization.
We also examined the intracellular localization of the different myc-tagged Vps26 constructs by immunofluorescence microscopy. We observed that whereas the wild-type and R69A E71A Vps26 constructs colocalized with endogenous SNX1 on endosomes, the I235D M236D, Δ238–246 GG, 238–246 polyS and G238P Vps26 constructs showed a diffuse cytosolic distribution ( Fig. 6b). These results agree with those of the co-precipitation experiments and additionally demonstrate that recruitment of Vps26 to endosomes requires its assembly with Vps29 and Vps35.
Next, we investigated the effect of Vps26 mutations on retromer function in S. cerevisiae. Yeast Vps26 was tagged at its C-terminus with GFP, and mutations were constructed that were analogous to those previously made on human Vps26. The resulting Vps26-GFP constructs were expressed by transformation into a Vps26-null yeast strain (vps26Δ ). The transformed strains were then examined for expression of the different Vps26 constructs, and for processing and secretion of CPY. Immunoblot analysis showed that all the Vps26 constructs were expressed at similar levels ( Fig. 7a). In vps26Δ cells transformed with wild-type Vps26-GFP, most of the Golgi precursor (p2) form of CPY was processed to the mature (m) form, as determined by metabolic-labeling, pulse-chase analysis (Fig. 7b, lane 1). In contrast, vps26Δ strains transformed with GFP-tagged I318D M319D, Δ321–328 GG, 321–328 polyS, or G321P, or GFP alone exhibited a marked CPY processing defect as indicated by the accumulation of the p2 form of CPY (Fig. 7, lanes 2–6). A colony-blotting assay similarly showed that vps26Δ cells expressing I318D M319D, Δ321–328 GG, 321–328 polyS or G321P Vps26 constructs tagged with GFP, or GFP alone, secreted higher amounts of CPY relative to that secreted by vps26Δ cells expressing wild-type Vps26-GFP ( Fig. 7c). From these experiments, we concluded that the ability of Vps26 to be incorporated into the retromer complex is essential for retromer function
Figure 7. Analysis of CPY sorting in wild-type and mutant Vps26p-expressing yeast strains.
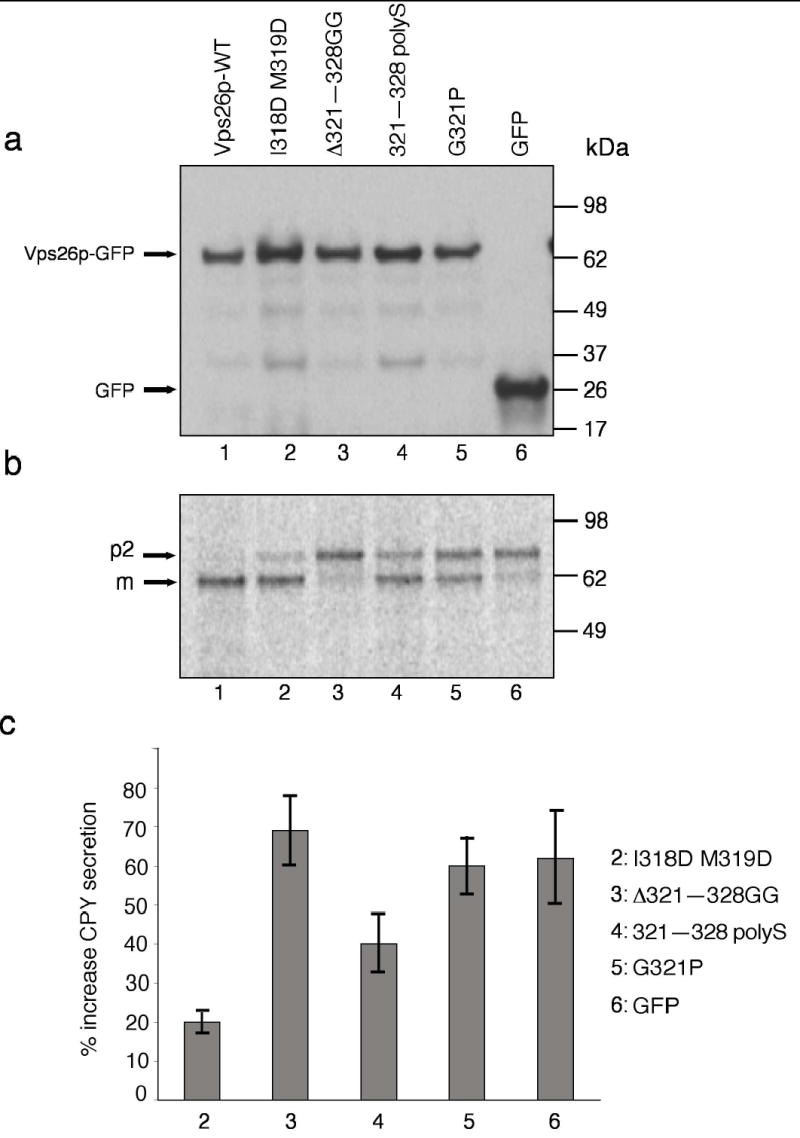
(a) S. cerevisiae vps26Δ strains transformed with expression plasmids encoding wild-type (lane 1) or I318D M319D (lane 2), Δ321–328 GG lane3, 321–328 polyS (lane 4) or G321P (lane 5) Vps26-GFP constructs, or GFP (lane 6), were lysed and analyzed by SDS-PAGE and immunoblotting using anti-GFP antibody. (b) The processing of CPY in the same strains described in (a) was analyzed by Metabolic-labeling, pulse-chase analysis and immunoprecipitation with antibody to CPY. Lanes correspond to the same constructs mentioned in (a). The positions of the Golgi precursor (p2) and mature (m) forms of CPY are indicated (c) Secretion of CPY into the medium by S. cerevisiae vps26Δ strains transformed with expression plasmids encoding the mutant Vps26-GFP constructs I318D M319D, Δ321–328 GG, 321–318 polyS and G321P, or GFP, was analyzed using a colony blotting assay. Values represent the percent increase of CPY secretion relative to the CPY secreted by a wild-type-Vps26-GFP-transformed vps26Δ strain. Results are expressed as mean ± SEM from four independent experiments.
Finally, we investigated the functional consequences of mutating residues in the polar core of Vps26 (i.e., Y121F/Y150F, N125R/N155D, E215R/E298R, E215R/E298R, in human/yeast; Table 1) on its recruitment to endosomes in human cells and rescue of CPY secretion in vps26Δ yeast cells. We found that these mutants behaved like wild-type Vps26 in both assays (Table 1 and data not shown). These results are consistent with the fact that incorporation of Vps26 into the retromer complex is mediated by the distal tip of the C-terminal domain, and not the polar core, of Vps26. Additionally, these findings indicate that single mutations in the polar core of yeast Vps26 do not abrogate its function in CPY sorting.
DISCUSSION
The first major observation in this study is the completely unexpected structural similarity between the retromer subunit Vps26 and the arrestin family of trafficking proteins. Vps26 and the arrestins have in common an unusual polar core that links the two β-sandwich domains. The arrestins have emerged as an important class of trafficking adaptor proteins 28,34. Arrestins directly bind to clathrin, the adaptor protein complex AP- 2, the ubiquitin ligase Mdm2, the small GTPase ARF6, the nucleotide exchange factor ARNO, the plasma membrane phospholipid PI(4,5)P2, and a range of receptors and other signaling proteins 28. The clathrin, phosphopeptide, ubiquitination, and PI(4,5)P2 binding sites found in the arrestins are absent in Vps26, however. Although the receptor-binding cups in the N and C domains of arrestin correspond to cups of a very similar shape and polarity in Vps26, the detailed nature of the residues is not conserved. Two yeast dominant-negative mutants of unknown biochemical effect 33 map to the N domain cup, which corresponds to part of the G-protein coupled receptor binding site in the arrestins. These two residues are similar in human and other Vps26 orthologs, suggesting that at least the N domain cup has an important function in Vps26.
The inactive conformations of visual arrestin 31,35 and β-arrestin 36,37 are locked in place by interactions between the interdomain linker, the C-terminal loop, and the polar core. The active conformation of arrestin has been inferred from mutational 38 and proteolytic 39 analyses. Upon receptor phosphorylation, the phospho-Ser in the receptor tail binds to Arg175 in the polar core. This destabilizes the polar core such that the two β-sandwich domains can reorient with respect to each other and embrace the GPCR through their concave surfaces 40. Visual arrestin Arg175 is replaced by hydrophobic residues in Vps26 orthologs, suggesting no arrestin-like phosphate sensor in the Vps26 family. We have mutated four polar core residues in Vps26, and these mutations do not interfere with endosomal localization in human cells or CPY sorting in yeast, so the functional significance of the polar core in Vps26 is currently unclear.
The second major observation in this study is that Vps26 binds to Vps35 and is integrated into the retromer complex via an exposed surface loop near the tip of the C-terminal domain. Since the Vps35 binding site involves only the tip of one domain, Vps35 interactions will be independent of any rearrangements of the two Vps26 domains relative to each other. This would allow Vps26 to undergo a regulatory conformational cycle whilst remaining integrated in the complex. The importance of this loop is highlighted by its conservation in Vps26 orthologs and paralogs. A second Vps26 isoform, Vps26B, was recently described 27 and reported to localize to the plasma membrane, rather than endosomes. Since this loop contains the determinants for the endosomal localization of Vps26A, it will be interesting to understand the mechanism for the apparently distinct localization of Vps26B. The loop is also conserved in yeast Vps26. We have found an exquisite correlation between loss of human Vps35 binding and endosomal localization in tissue culture cells and loss of CPY sorting function in yeast.
The determinants for Vps35 binding include two hydrophobic residues and a Gly residue. The key role of a Gly residue suggests that the loop binds to a well-defined pocket on the surface of Vps35 that requires an unusual conformation of the loop, is sterically tightly packed, or both. One precedent for such a role for a Gly residue is illustrated by the GGA appendage binding motifs of rabaptin-5 and related proteins 41,42. The key functional role of the two hydrophobic residues suggests a hydrophobic character for at least part of the pocket on Vps35.
Vps26 has been the most mysterious player of the five retromer subunits because of the absence of recognizable homology to any other protein of known structure or function and the lack of identified interactions with other proteins. The findings in this study now provide a structural foothold for understanding the function of Vps26 orthologs. We now know that Vps26 has a common architecture with the arrestins. The similarities are extensive enough to suggest that Vps26 undergoes an arrestin-like conformational change that enables it to bind transmembrane receptors of some kind. It will be important to test this idea and to seek the identity of the putative receptors that interact with Vps26 orthologs and the factors that control the putative conformational change. While these concepts of receptor binding and conformational change remain to be tested, the observations in this study provide compelling evidence for a mechanism by which Vps26 orthologs are assembled into the retromer.. We have found that the Vps35 binding site on Vps26 is essential for localization in mammalian cells and for function in yeast, and it seems likely that these findings will be general across other species.
METHODS
Protein expression, purification, and crystallization
PCR was used to amplify the cDNA coding for human Vps26A and to add a C-terminal His5 tag (GLVPRGSHHHHH). The product was inserted into the pmr101A (modified pmr101; American Type Culture Collection, Manassas, Virginia) vector using the NdeI and XhoI restriction sites. The expressed recombinant protein contains the additional dipeptide MetGly at the N terminus as a result of the vector construction. Protein was expressed in the BL21 (DE3) star (Invitrogen) strain at 20°C overnight. Vps26 was purified on a 5 ml HiTrap Chelating HP column (GE Healthcare) and eluted with a gradient of 12.5 mM to 300 mM imidazole in a buffer containing 20 mM Tris pH 8.0, 50 mM NaCl, 5% (v/v) glycerol. The eluted fractions were pooled, diluted by adding an equal volume of water, and purified by ion exchange (5 ml HiTrap Q FF) and gel filtration (Superdex 75 16/60) (GE Healthcare). Fractions were pooled and concentrated to 8 mg ml−1 in 10 mM Tris pH 8.0, 5% (v/v) glycerol, 100 mM NaCl, and 10 mM dithiothreitol (DTT) for crystallization.
Diffraction quality crystals used for native data collection were obtained using buffer containing 100 mM Na cacodylate pH 6.5, 15% (v/v) glycerol, 8% (w/v) PEG 10,000, 10 mM DTT and 1 mM TmCl3 by micro-seeding. SeMet substituted protein was produced using the same strain growing in media containing 1g NH4Cl, 3g KH2PO4, 6g Na2HPO4·7H2O, 2.5g NaAc·3H2O, 1.5 g succinic acid, 20 g glucose, 500 mg MgSO4·7H2O, 13.9 mg FeSO4·7H2O, 10 ml Kao and Michayluk vitamin solution (100×) (Sigma) per liter, and the pH was adjusted to 7.4 with NaOH. D,L-SeMet was added to the media when the cells had grown to OD600 = 0.5, to a final concentration of 100 μg/ml. The culture was induced 20 minutes after the addition of SeMet, and harvested after overnight incubation at 20°C. The expression, purification and crystallization of the Se derivative protein were carried under the same conditions as that of native protein, except that GdCl3 was used as a crystallization additive instead of TmCl3. A Pt derivative was prepared by soaking native crystals in buffer containing 100 mM Na cacodylate pH 6.5, 20% (v/v) glycerol, 8% (w/v) PEG 10,000 and 10 mM Pt(NH3) 2Cl2 at 23 °C for five days.
Data collection and structure determination
Data were collected at 98o K at beamline ID-22, Advanced Photon Source (Argonne National Lab, IL; Table 2) and processed with HKL2000 (HKL Research). The structure was determined by combining a two wavelength MAD data set from a SeMet crystal, a three wavelength MAD data set from a Pt crystal, and an SIR analysis using the Pt crystal as a derivative. Six ordered Se and one Pt site were identified using SOLVE 43. Subsequent phasing and solvent flattening was carried out using SOLVE43 and RESOLVE 44. The solved structure revealed electron density attached to the two Cys residues of Vps26, which we attribute to covalently bound dimethylarsenate, which was included in the refined model. Iterative cycles of model building and refinement were carried out using the O 45, CNS 46 and CCP4 47 program packages. 8.8% of the data was used for cross-validation in CNS.
Table 2.
Data collection, phasing and refinement statistics
| Native | Se | Pt | ||||
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P212121 | P212121 | ||||
| Cell dimensions | ||||||
| a, b, c (Å) | 41.0, 76.7, | 41.14, 77.06, | 40.6, 76.2 | |||
| 126.5 | 126.8 | 128.4 | ||||
| α, β,γ (°) | 90 90 90 | 90 90 90 | 90 90 90 | |||
| Inflection | Remote | Peak | Inflection | Remote | ||
| Wavelength | 1.072 Å | 0.9794 Å | 0.9686 Å | 1.072 Å | 1.054 Å | 1.5418 Å |
| Resolution (Å) | 2.1 | 3.0 | 3.0 | 2.5 | 2.7 | 2.6 |
| Rsym or Rmerge | 4.3 (34.7) | 7.5 (14.1) | 7.6 (10.1) | 8.1 (21.9) | 6.9 (22.8) | 6.9 (50.4) |
| I/σI | 21.3 | 16.0 | 13.8 | 13.4 | 14 | 18.3 |
| Completeness (%) | 92.2 (81.8) | 96.5 (89.1) | 97.4 (75.6) | 98.5 (96.1) | 98.9 (97.0) | 98.1 (87.5) |
| Redundancy | 7 | 10 | 11 | 8 | 4 | 13 |
| Refinement | ||||||
| Resolution (Å) | 2.1 | |||||
| No. reflections | 21275 | |||||
| Rwork/Rfree | 23.4/28.5 | |||||
| No. atoms | ||||||
| Protein | 2380 | |||||
| Ligand/ion | 6 | |||||
| Water | 89 | |||||
| B-factors | ||||||
| Protein | 65.765 | |||||
| Ligand/ion | 75.9 | |||||
| Water | 63.85 | |||||
| R.m.s deviations | ||||||
| Bond lengths (Å) | 0.008 | |||||
| Bond angles (°) | 1.37 | |||||
Three crystals are used for structure solving and refinement *Highest-resolution shell is shown in parentheses.
Recombinant DNA constructs
An EcoRI-XhoI fragment encoding human Vps26A (residues 2–327) obtained from the Vps26A-pB42AD construct 32 was subcloned into the corresponding sites of the pGADT7 vector (Clontech, Mountain View, CA) and the pcDNATM3.1/myc-His vector (Invitrogen, Carlsbad, CA). Addition of a start codon (position 1) and removal of a stop codon (position 327) from the Vps26A-pcDNA3.1 construct was done by site-directed mutagenesis. A MfeI/BamHI fragment encoding Vps35 (residues 2–796) was obtained from the Vps35-pLexA construct 32 and later subcloned into the EcoRI and BamHI sites of the pGBKT7 vector (Clontech). An EcoRI/Bamhi fragment encoding full-length S. cerevisiae Vps26p was cloned into the pRS316 (C-terminal GFP) vector. Mutagenesis of the Vps26A-pGADT7, Vps26A-myc-pcDNA3.1, and the Vps26p-pRS316 constructs was performed by using the QuickChange Site-Directed Mutagenesis kit (Stratagene). All the constructs were sequenced to confirm their identity and the presence of the desired mutations.
Yeast two-hybrid assay
The S. cerevisiae strain AH-109 (Clontech) was transformed by the lithium acetate procedure. Yeast cells were co-transformed with cDNAs encoding wild-type Vps35 fused to Gal4BD (Vps35-pGBKT7) and wild-type (WT) or mutant Vps26A fused to GAL4AD (Vps26-pGADT7). After selection, the co-transformants were spotted on plates containing (+His) or lacking (-His) histidine. As negative controls, the AH-109 strain was co-transformed with Vps26- pGADT7 and pGBKT7, Vps35-pGBKT7 and pGADT7, and the “empty” vectors pGBKT7 and pGADT7. As a positive control for growth on –His plates, cells were co-transformed with p53-pGBKT7 and SV40-LT-Ag-pGADT7 (Tag). AH-109 transformants were grown overnight at 30°C in media lacking leucine and tryptophan. The day of the experiment, the yeast cell cultures were diluted to 0.4 OD600 and grown for 2h at 30°C. The cells were then harvested at 2500 rpm, washed and resuspended in medium lacking leucine and tryptophan to 0.1 OD600/ml. Five μl of each suspension was spotted on plates lacking leucine and tryptophan, in the presence or absence of histidine, and incubated at 30°C for 2–3 days. Whole cell lysates of the transformants were obtained according to published protocols 48 and subjected to SDS-PAGE and immunoblot analysis.
Immunoprecipitation
HeLa cells (American Type Culture Collection) were cultured on 100 mm dishes as described previously 49. When the cells reached 80–90% confluency, they were transfected with 24 μg of pcDNA3.1 encoding myc-tagged forms of wild-type, I235D M236D, Δ238–246 GG, 238–246 polyS, G238P, R69A E71A Vps26 using Lipofectamine-2000 (Invitrogen, Carlsbad, CA). At 24 h after transfection, cells were washed twice with ice-cold PBS and immediately resuspended in 1 ml lysis buffer (0.5 % (v/v) Triton X-100, 300 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA) supplemented with a protease inhibitor cocktail (Roche, Indianapolis, IN). After 30 min of incubation at 4°C, lysates were centrifuged at 16,000 g for 15 min. The supernatants were then pre-cleared by incubation for 60 min at 4°C with 30 μl Protein G-Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ) and centrifugation at 8,000 g for 5 min. The pre-cleared lysates were subsequently incubated for 2 h at 4°C with 30 μl Protein G-Sepharose beads bound to mouse monoclonal anti-myc (9E10) antibody (Covance, Princeton, NJ). Following immunoprecipitation, the beads were washed four times with ice-cold wash buffer (0.1 % (w/v) Triton X-100, 300 mM NaCl, 50 mM Tris-HCl pH 7.4) and once with ice-cold PBS. Washed beads were subsequently subjected to SDS-PAGE and immunoblotting analysis. Antibodies to Vps29 and Vps35 have been described before 32.
Immunofluorescent labeling and scanning-laser confocal microscopy
HeLa cells grown on glass cover slips and transfected with wild-type and mutant Vps26A-myc-pcDNA3.1constructs were washed with PBS and fixed for 10 min with 4% (w/v) paraformaldehyde in PBS at room temperature. Cells were rinsed twice with PBS, and excess paraformaldehyde was quenched with PBS containing 20 mM glycine, pH 8.0, and 75 mM NH4Cl for 15 min at room temperature. The cells were again washed with PBS and permeabilized with 0.025% (w/v) saponin prepared in blocking solution (PBS containing 5% v/v goat serum and 7 mg ml−1 fish skin gelatin) for 10 min at 37°C in a humidified chamber. Cells were immunostained with mouse monoclonal anti-myc (9E10) antibody and polyclonal anti-SNX1 for 2h at 37°C, followed by incubation with fluorescently-labeled secondary antibodies. Imaging was performed on a TCS SP-2 confocal microscope (Leica, Deerfield, IL) equipped with argon/krypton, and 543/594 nm helium-neon lasers. Images were acquired using a 63x Plan-Apochromat oil objective (NA 1.4) and the appropriate filter combination. Settings were as follows: photomultipliers set to 500–700 V, Airy = 1, zoom = 3.0–4.0, Kalman filter (n=8). The images (512 x 512 pixels) were saved in a tag-information-file-format (TIFF), contrast was corrected in Photoshop (Adobe, San Jose, CA), and images were imported into Freehand MX (Macromedia, San Francisco, CA).
Yeast growth and CPY sorting assays
The Vps26Δ::KanMX deletion S. cerevisiae strain (Vps26Δ) was obtained from Research Genetics (Invitrogen, Carlsbad, CA) and its ability to sort and process CPY was compared with the parental strain BY4742 (data not shown). Yeast cells were maintained in yeast-peptone-dextrose (YPD) plates or grown in liquid medium before transformation with yeast expression plasmids. Transformation with pRS316 encoding GFP-tagged forms of wild-type, I318D M319D, Δ321–328 GG, 321–328 polyS, G321P Vps26 or GFP into the VPS26Δ strain was performed by the lithium acetate procedure.
Wild-type and mutant Vps26p-GFP-pRS316-transformed cells were metabolically labeled with the 35S Express reagent (Perkin Elmer Life Sciences) for 10 min (pulse) and chased for 30 min as previously described 48. Immunoprecipitations using mouse-anti-CPY (Molecular Probes, Eugene, OR) conjugated to Protein G-Sepharose beads were performed overnight at 4°C as previously described 48 and the immunoprecipitates were analyzed by SDS-PAGE and autoradiography. Dried gels were also scanned on a Phosphoimager (Typhoon 9200, Molecular Dynamics, Sunnyvale, CA).
The CPY colony blot assay was adapted from ref. 50. Yeast transformants grown in selection medium were spotted (1.0x10−3 OD600 of cells) onto a YPD plate and overlaid with a nitrocellulose membrane. The plates were incubated for 22–24 h at 30°C. The nitrocellulose membranes were washed with water and immunoblotted with mouse anti-CPY and the appropriate HRP-conjugated secondary antibody. Visualization was done with enhanced chemiluminescence reagents (Perkin Elmer Life Sciences, Boston, MA).
Coordinates
The crystallographic coordinates have been deposited in the protein data bank with accession code 2FAU.
Supplementary Material
Acknowledgments
We thank B. Beach and X. Zhu for technical assistance, W. Smith for liposome binding studies, H. Watson and C. Bonangelino for advice on CPY sorting assays, C. Haft for reagents and comments on the manuscript, and D. Hurt, G. Miller, and the staff of beamline 22-ID, Advanced Photon Source (APS), Argonne National Laboratory (ANL) for assistance with X-ray data collection. This research was supported by the NIH intramural programs of the NIDDK (to J. H. H.) and the NICHD (to J. S. B.). Use of the APS was supported by the U. S. DOE, Basic Energy Sciences, Office of Science, under Contract No.W-31-109-Eng-38.
References
- 1.Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: New twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 2.Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- 3.Meyer C, et al. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. Embo J. 2000;19:2193–2203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doray B, Ghosh P, Griffith J, Geuze HJ, Kornfeld S. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science. 2002;297:1700–1703. doi: 10.1126/science.1075327. [DOI] [PubMed] [Google Scholar]
- 5.Diaz E, Pfeffer SR. TIP47: A cargo selection device for mannose 6-phosphate receptor trafficking. Cell. 1998;93:433–443. doi: 10.1016/s0092-8674(00)81171-x. [DOI] [PubMed] [Google Scholar]
- 6.Wan L, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94:205–216. doi: 10.1016/s0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- 7.Bowers K, Stevens TH. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochimica Et Biophysica Acta-Molecular Cell Research. 2005;1744:438–454. doi: 10.1016/j.bbamcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Seaman MN. Recycle your receptors with retromer. Trends Cell Biol. 2005;15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Horazdovsky BF, et al. A sorting nexin-1 homologue, vps5p, forms a complex with vps17p and is required for recycling the vacuolar protein-sorting receptor. Mol Biol Cell. 1997;8:1529–1541. doi: 10.1091/mbc.8.8.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seaman MNJ, McCaffery JM, Emr SD. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol. 1998;142:665–681. doi: 10.1083/jcb.142.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seaman MNJ, Williams HP. Identification of the functional domains of yeast sorting nexins Vps5p and Vps17p. Mol Biol Cell. 2002;13:2826–2840. doi: 10.1091/mbc.02-05-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheever ML, et al. Phox domain interaction with Ptdlns(S)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- 13.Kanai F, et al. The PX domains of p47phox and p40phox bind to lipid products of Pl(3)K. Nat Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Hortsman H, Seet L, Wong SH, Hong W. SNX3 regulates endosomal function through its PX-domain-mediated interaction with PtdIns(3)P. Nat Cell Biol. 2001;3:658–66. doi: 10.1038/35083051. [DOI] [PubMed] [Google Scholar]
- 15.Ellson CD, et al. Ptdlns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox) Nat Cell Biol. 2001;3:679–682. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 16.Virbasius JV, et al. Activation of the Akt-related cytokine-independent survival kinase requires interaction of its phox domain with endosomal phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2001;98:12908–12913. doi: 10.1073/pnas.221352898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peter BJ, et al. BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 18.Nothwehr SF, Bruinsma P, Strawn LA. Distinct domains within Vps35p mediate the retrieval of two different cargo proteins from the yeast prevacuolar/endosomal compartment. Mol Biol Cell. 1999;10:875–890. doi: 10.1091/mbc.10.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. 2004;165:123–33. doi: 10.1083/jcb.200312055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlton J, et al. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high-curvature membranes and 3-phosphoinositides. Curr Biol. 2004;14:1791–1800. doi: 10.1016/j.cub.2004.09.077. [DOI] [PubMed] [Google Scholar]
- 21.Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–22. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Q, et al. Determinants of the endosomal localization of sorting nexin 1. Mol Biol Cell. 2005;16:2049–57. doi: 10.1091/mbc.E04-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, et al. Crystal Structure of Human Vacuolar Protein Sorting Protein 29 Reveals a Phosphodiesterase/Nuclease-like Fold and Two Protein-Protein Interaction Sites. J Biol Chem. 2005;280:22962–7. doi: 10.1074/jbc.M500464200. [DOI] [PubMed] [Google Scholar]
- 24.Collins BM, Skinner CF, Watson PJ, Seaman MN, Owen DJ. Vps29 has a phosphoesterase fold that acts as a protein interaction scaffold for retromer assembly. Nat Struct Mol Biol. 2005 doi: 10.1038/nsmb954. [DOI] [PubMed] [Google Scholar]
- 25.Lee JJ, Radice G, Perkins CP, Costantini F. Identification and characterization of a novel, evolutionarily conserved gene disrupted by the murine Hb58 embryonic lethal insertion. Development. 1992;115:227–288. doi: 10.1242/dev.115.1.277. [DOI] [PubMed] [Google Scholar]
- 26.Small SA, et al. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Annals Of Neurology. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 27.Kerr MC, et al. A novel mammalian retromer component, Vps26B. Traffic. 2005;6:991–1001. doi: 10.1111/j.1600-0854.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 28.Lefkowitz RJ, Whalen EJ. beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Hurley JH, Misra S. Signaling and subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomolec Struct. 2000;29:49–79. doi: 10.1146/annurev.biophys.29.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm L, Sander C. Dali - a Network Tool for Protein-Structure Comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 angstrom crystal structure of visual arrestin: A model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 32.Haft CR, et al. Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: Assembly into multimeric complexes. Mol Biol Cell. 2000;11:4105–4116. doi: 10.1091/mbc.11.12.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy JV, Seaman MN. Vps26p, a component of retromer, directs the interactions of Vps35p in endosome-to-Golgi retrieval. Mol Biol Cell. 2001;12:3242–56. doi: 10.1091/mbc.12.10.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodman OB, et al. beta-arrestin acts as a clathrin adaptor in endocytosis of the beta(2)-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 35.Granzin J, et al. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 1998;391:918–921. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- 36.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 angstrom: Possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 37.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 38.Vishnivetskiy SA, Hirsch JA, Velez MG, Gurevich YV, Gurevich VV. Transition of arrestin into the active receptor-binding state requires an extended interdomain hinge. J Biol Chem. 2002;277:43961–43967. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- 39.Xiao KH, Shenoy SK, Nobles K, Lefkowitz RJ. Activation-dependent conformational changes in beta-arrestin 2. J Biol Chem. 2004;279:55744–55753. doi: 10.1074/jbc.M409785200. [DOI] [PubMed] [Google Scholar]
- 40.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface - Structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 41.Miller GJ, Mattera R, Bonifacino JS, Hurley JH. Recognition of accessory protein motifs by the gamma-adaptin ear domain of GGA3. Nat Struct Biol. 2003;10:599–606. doi: 10.1038/nsb953. [DOI] [PubMed] [Google Scholar]
- 42.Collins BM, Praefcke GJK, Robinson MS, Owen DJ. Structural basis for binding of accessory proteins by the appendage domain of GGAs. Nat Struct Biol. 2003;10:607–613. doi: 10.1038/nsb955. [DOI] [PubMed] [Google Scholar]
- 43.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr Sect D-Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr Sect D-Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved Methods for Building Protein Models in Electron- Density Maps and the Location of Errors in These Models. Acta Crystallogr Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 46.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D-Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect A. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 48.Bonifacino JS, Dell'Angelica EC. Current Protocols in Cell Biology. In: Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. John Wiley and Sons, Inc.; New York, NY: Protein labeling and immunoprecipitation. 1998. [Google Scholar]
- 49.Mattera R, Arighi CN, Lodge R, Zerial M, Bonifacino JS. Divalent interaction of the GGAs with the Rabaptin-5-Rabex-5 complex. Embo J. 2003;22:78–88. doi: 10.1093/emboj/cdg015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonangelino CJ, Chavez EM, Bonifacino JS. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2486–2501. doi: 10.1091/mbc.02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.