

Abstract
The funnel landscape model predicts that protein folding proceeds through multiple kinetic pathways. Experimental evidence is presented for more than one such pathway in the folding dynamics of a globular protein, cytochrome c. After photodissociation of CO from the partially denatured ferrous protein, fast time-resolved CD spectroscopy shows a submillisecond folding process that is complete in ≈10−6 s, concomitant with heme binding of a methionine residue. Kinetic modeling of time-resolved magnetic circular dichroism data further provides strong evidence that a 50-μs heme–histidine binding process proceeds in parallel with the faster pathway, implying that Met and His binding occur in different conformational ensembles of the protein, i.e., along respective ultrafast (microseconds) and fast (milliseconds) folding pathways. This kinetic heterogeneity appears to be intrinsic to the diffusional nature of early folding dynamics on the energy landscape, as opposed to the late-time heterogeneity associated with nonnative heme ligation and proline isomers in cytochrome c.
Extensive experimental and theoretical efforts are underway to understand how proteins fold spontaneously to their native functional conformations. Much effort has been focused on models posed in the chemical kinetic form traditional for small molecules, i.e., in terms of a mechanistic pathway involving a number of intermediates linked by passages over intervening transition states (1–3). However, this approach may be inadequate to describe the complex interactions expected to shape the course of protein folding. Another approach explicitly recognizes the statistical dimensions of the folding problem and considers ensembles of conformations evolving toward the native state by biased diffusion over a rough energy landscape (4–7). Escape from the Levinthal paradox (8), which suggests that a protein randomly searching all of its configurational space will never fold on physiological time scales, is afforded in this picture by a global bias in the landscape, a folding “funnel,” guiding diffusion between nonnative configurational traps toward the native configuration (9). This proposal suggests that amino acid sequences observed to fold do so because they have a single funnel on their energy landscape, whereas nonfoldable sequences have too many funnels and no unique native state. This view also implies that folding intermediates with different conformations may be kinetically inaccessible to one another if diffusion between them is slower than diffusion down the funnel(s), in which case the intermediates lie on distinct pathways. In principle, these pathways may be very great in number, although a molecular dynamics simulation of unfolding in a small protein, chymotrypsin inhibitor 2, suggests that a small number of statistically preferred pathways may exist on a funnel-like average energy surface (10).
The strongest impetus for the funnel landscape view has come from theoretical studies, e.g., lattice simulations (11–14) and statistical mechanical models (5, 11, 15), although the results of steady-state partial denaturation and kinetic experiments also offer some empirical support for this view (16, 17). In particular, the landscape model predicts a nonsequence-specific relationship between the rate constant for type I folding (entropic-bottleneck limited) and the free energy of folding that corresponds to that observed for the millisecond photoreduction-induced folding reactions of cytochrome c obtained from different species (16, 18).
Multiple pathways have been reported previously for the millisecond and longer folding reactions of a number of proteins (19). However, in contrast to the multiple pathways predicted by the energy landscape model, which arise in that picture from the diffusional nature of the folding process, the cause of kinetic heterogeneity can be traced in nearly all observed cases to the presence of nonnative ligation or proline isomers. The folding of bovine pancreatic trypsin inhibitor, for instance, goes through two pathways distinguished by different patterns of disulfide bond formation (20, 21). Similarly, slow isomerization of the prolyl peptide bond has been identified with separate slow-folding phases in many proteins, including cytochrome c (22). The formation of nonnative heme-residue contacts is known to be a further source of multiple folding phases on the millisecond time scale in cytochrome c (23). [Kinetic heterogeneity has also been observed in the folding reactions of some proteins, such as dihydrofolate reductase (24) and hen egg white lysozyme (25), apparently lacking slow isomerization or ligation steps.] The kinetic heterogeneity associated with folding traps (type II, very rough energy-landscape folding) is expected to be most important in the late folding events of proteins (16). This view suggests that investigations of multiple pathways intrinsic to the diffusional nature of the energy landscape model are best directed toward early folding events expected to conform to type 0 (downhill) or type I scenarios (16).
Because triggering with light offers the possibility of resolving the earliest folding processes in time, fast optical triggers such as IR-laser temperature jump (26), chromophore photoisomerization, electron photoinjection (27), and ligand photodissociation (28) have been used to induce folding and unfolding reactions (16, 29). We used the photoinitiation mechanism of Jones et al. (28), photodissociation of the CO complex of cytochrome c partially denatured in guanidine HCl (GuHCl). Photolysis is expected to trigger folding because the protein is destabilized by displacement of the native ligand, Met-80, by exogenous CO at the heme Fe(II) axial coordination site.
We report here the results of fast time-resolved circular dichroism (TRCD) and magnetic circular dichroism (TRMCD) measurements for reduced cytochrome c after photodissociation of the denatured CO complex of this globular 12-kDa protein. These measurements reveal a heterogeneity in the kinetics of early folding events, as determined by kinetic modeling of the TRMCD data, consistent with the dynamics for an ensemble of pathways on the energy landscape. The TRCD measurements, described in detail elsewhere (30), find a small submillisecond folding component that corresponds roughly in amplitude and kinetics with the 2-μs appearance of a native heme-ligation species previously observed with absorption spectroscopy (28). TRMCD measurements were undertaken to investigate this correspondence, which suggests that the submillisecond folding is global within a distinct subset of the sample rather than subdomain folding throughout the sample. Distinct folding ensembles were detected in the present study by the magnetic circular dichroism (MCD) spectral signatures associated with either a methionine or a histidine residue as the sixth axial heme ligand. Kinetic heterogeneity appearing before the heme has had time to form the nonnative ligation intermediates known to give rise to multiphase folding kinetics at later times (31) most probably reflects the influence of intrachain diffusional processes in limiting access of methionine and histidine residues to the heme binding site. To the extent that our TRMCD results support the existence of such early-time heterogeneity in reduced cytochrome c, we interpret this time-resolved spectral information as providing the first direct experimental support for the multiple kinetic pathway prediction of the biased-diffusion landscape model of protein folding.
MATERIALS AND METHODS
Materials.
Horse-heart cytochrome c (Sigma, purity >99%) was partially denatured in 4.6 M GuHCl, pH 6.5 phosphate buffer and saturated with 1 atm (101.3 kPa) of CO to produce nearly quantitative CO complex.
TRCD.
A nanosecond laser photolysis apparatus modified for TRCD used a near-null ellipsometric method with a reference strain birefringence (δ) of 2.0° (30, 32). Ten thousand spectral scans were averaged for 106-μM samples, 0.05 cm pathlength, at 40°C. Fresh sample was flowed into the cell between scans to remove laser photodegradation products. Photolysis was initiated by a Q-switched Nd:yttrium/aluminum garnet laser generating 7-ns 26-mJ pulses of 532-nm second harmonic light at a repetition rate of 2 Hz. The broad-band probe beam from a microsecond xenon flash lamp was collected after the sample and detected with a spectrograph and optical multichannel analyzer.
TRMCD.
Soret TRMCD spectra were obtained from the Kramers–Kronig transform of time-resolved magnetic optical rotatory dispersions averaged from 1,300 scans measured on a modified ns laser photolysis apparatus by using a near-null polarimetric technique with a reference polarizer rotation angle (β) of 620 mdeg (32, 33). The sample was prepared as described for the far-UV TRCD data, with a protein concentration of 94 μM, and magnetized by a 1.0-T electromagnet. Sample excitation and data collection were the same as in the TRCD measurements except that the excitation and probe beams were collinear instead of at an angle of 30°.
Singular Value Decomposition (SVD) and Global Kinetic Analysis.
The TRMCD spectra were filtered of experimental noise by SVD and then globally analyzed for multiexponential kinetics. The filtered data matrix Ar included only the SVD components corresponding to the rth largest singular values, r being the effective rank of the data matrix (r = 4 for the TRMCD data). Nonlinear fitting of Ar = BT yielded exponential time (T) and spectral (B, or “b-spectra”) functions associated with each observed process. Details of SVD and global kinetic analysis have been reviewed (34, 35).
The kinetic models considered in this work contain first-order or pseudo-first-order processes and can be written symbolically in matrix form as [dc(t)/dt] = Kc(t), where K is the matrix of rate constants and c(t) is a column vector of time-dependent concentrations. A general solution for the concentrations is
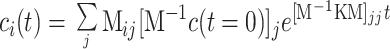 |
1 |
where K is diagonalized by a similarity transform with the matrix M. The observed time constants found above correspond to the diagonal elements of M−1KM, which we will call Kobs. In the simplest parallel process model, the model used in finding the observed time constants and b-spectra above, K = Kobs, (M = identity matrix), and the columns of T correspond to the ci(t) at the indexed times, with ci(t = 0) = 1. For the more complex heterogeneous kinetic model considered below, M was calculated from M−1KM = Kobs and the ci(t = 0) were determined by optimizing the similarity of the MCD spectra calculated for the intermediates to the spectra of model compounds. This optimization used two free parameters corresponding to the initial fractional concentrations of two of the three ensemble populations, the concentration of the third ensemble being fixed by the constraint that the fractional concentrations sum to unity. The matrix of intermediate spectra, F, was calculated from F = Arpinv(C), where pinv(C) is the matrix pseudoinverse of C, the matrix of intermediate concentrations. Finally, a homogeneous multiple-equilibria kinetic model is also considered below. It contains two parameters as well, in this case finite equilibrium constants for Met and His binding to heme, that are not fixed by the observed time constants. As in the heterogeneous model, these free parameters were determined by optimizing the fit of intermediate spectra to the model spectra.
RESULTS AND DISCUSSION
We used nanosecond TRCD spectroscopy (32) of the amide bands to assess directly the extent of secondary structure change associated with submillisecond protein folding in this system, as described in more detail by Chen et al. (30). Although the thermodynamic driving force for folding is such that at least 95% of non-CO-bound protein is folded at equilibrium (28), the extent of folding observed on the submillisecond time scale is far less. The far-UV TRCD data show that secondary structure corresponding to about 8% of the difference between the equilibrium secondary structure contents of CO-bound and five-coordinate ferrous cytochrome c in GuHCl is formed about 1 μs after photolysis. Although a previous study using tryptophan fluorescence found no evidence for submillisecond folding in this system (36), the signal-to-noise ratio obtained in that work was probably not high enough to detect the small ultrafast-folding component reported here. This evolution in TRCD signal is clearly associated with the formation of secondary structure, because the spectral shape of the TRCD difference signal near 220 nm is consistent with the CD spectrum of native cytochrome c secondary structure (30). The formation of some global secondary structure on this early time scale is consistent with the 50- to 500-ns time constants observed for localized secondary structure formation in apomyoglobin (26). Photoreduction (27) and stopped-flow denaturant dilution (31) studies of oxidized cytochrome c indicate that nearly all of the protein should fold to near-native conformation by about a second. However, CO rebinding restabilizes unfolded structures at times later than about a millisecond in the present study, setting an upper limit on the folding time scales investigated.
Incomplete folding after photodissociation in this system is also due at least in part to the rapid formation of nonnative heme-residue contacts, kinetic barriers that frustrate further folding before CO returns diffusively from solution. Microsecond intermediates bearing nonnative heme–histidine contacts were detected in the cytochrome c-CO system by Jones et al. (28) by using nanosecond time-resolved optical absorption (TROA). Nonnative histidine–heme conformers were also found to impede the later stages of oxidized cytochrome c refolding in stopped-flow denaturant dilution experiments that identified His-33 as the predominant ligand responsible for a 10−1 s time scale barrier to folding (31). Heterogeneous nonnative ligation by His-26 and His-33 gives rise to fast and slow millisecond folding phases, respectively, in the oxidized protein.
TRMCD spectroscopy (32) of the heme Soret bands, more sensitive to heme ligation and spin state than TROA, was used to monitor heme ligation events after CO photodissociation. Observed time constants of about 2 and 50 μs were assigned previously to Met and His coordination events, respectively, on the basis of Soret TROA measurements (28, 30). More definitive evidence for these Met−Fe−His and His−Fe−His species (the second axial ligand is presumably His-18 in both cases) is provided by our TRMCD results. The Soret MCD spectra of the diamagnetic Met–His (37) and bis-His (38) heme coordination species differ more sharply in bandshape and intensity than the corresponding absorption spectra, which are only slightly shifted in wavelength. Both MCD spectra are also easily distinguished from that of the paramagnetic five-coordinate species, which resembles deoxyhemoglobin (39). The TRMCD spectra observed after photodissociation of CO from cytochrome c were analyzed for the spectral changes associated with four observed exponential decay constants, shown in Fig. 1. Comparison of the associated decay spectra obtained for the first two rate processes with model difference spectra, generated from the MCD spectra of heme proteins containing known axial heme ligation, confirms the assignment of the first process to Met binding and the second to His binding.
Figure 1.

(A) TRMCD spectra of cytochrome c at logarithmic time intervals (330 ns to 25 ms, eight times/decade) after CO photolysis (arrows show change with time). Global kinetic analysis finds spectral changes (b-spectra) corresponding to four exponential time constants: (B) 3 μs, (C) 50 μs, (D) 300 μs, and (E) 700 μs (dark lines). The first b-spectrum closely resembles a (normalized) [five-coordinate heme − Met–His] model difference spectrum (B, light line), having a strong minimum near 415 nm that arises from the 413-nm lobe of the positive MCD A term of the native protein (37), consistent with the formation of native-like coordination. The second b-spectrum lacks this feature, resembling a [five-coordinate − bis-His] difference spectrum (C, light line) based on model spectra from cytochromes c3 (44) and b5 (38), and is assigned to the formation of nonnative histidine coordination. The final two CO pressure-dependent rate processes are dominantly CO recombinations (D and E).
The observed TRMCD changes were fit to two possible first-order kinetic models, results shown in Fig. 2, to establish more clearly the identity of the species in which heme-residue binding occurs. The analysis below shows that both early processes involve binding to high-spin five-coordinate species that are the immediate products of CO photodissociation. In particular, no net replacement of Met by His as a heme ligand is found to be associated with the second process. This finding rules out kinetic mechanisms in which Met is displaced by His in the second process, such as occurs in a multiple equilibria model previously used to fit less ligation-sensitive TROA data (28).
Figure 2.

Intermediate MCD spectra calculated from the heterogeneous kinetic mechanism in Fig. 3: (A) The aggregate five-coordinate species, cytMet* + cytHis* + cytU*, (B) cyt[Met–Fe–His], (C) cyt[His–Fe–His]*, and (D) cyt*-CO (A–D, dark lines). These were optimized to fit the spectra of corresponding model cytochromes (A–D, light lines); see text. Intermediate spectra calculated from a multiple-equilibria mechanism (Scheme S1) are shown for comparison (A–D, - - -) (These overlay the spectra from the heterogeneous mechanism in all panels except C.).
It is particularly significant with regard to inferring a kinetic mechanism for cytochrome c-CO photolysis that Met and His residues appear to compete for the same heme axial binding site but are associated with distinct observed time constants in TROA and TRMCD kinetic studies. The distinct TROA time constants could be accounted for in a homogeneous mechanism by considering back reactions in linked equilibria for Met and His binding (28), but such a mechanism is ruled out by the MCD results. The distinct time constants thus point to parallel processes.
We propose that this kinetic heterogeneity results from the unraveling of secondary and tertiary structure in GuHCl denaturant to produce a distribution of unfolded conformations. Within this distribution, the conformational subpopulations cytMet*, cytHis*, and cytU* are distinguished by the respective positioning of either a methionine, a histidine, or the lack of such a residue, for facile binding to heme after CO photodissociation (Fig. 3). Given the lack of stable tertiary structure in the unfolded state, this positioning is expected to be more or less consistent with that of a random polymer. Gaussian random coil statistics predict that the effective concentrations of the residues as potential heme ligands are proportional to n−3/2 in a denaturing solvent just sufficient for unfolding, n being the number of residues separating a given ligand from the heme site on the chain (40). This proportionality suggests that the effective histidine concentration will be several times higher than that of methionines.
Figure 3.

Heterogeneous kinetic model in which multiple folding pathways after photodissociation of partially denatured cytochrome c-CO (cyt*-CO) are distinguished by geminate positioning of different residues (Met, His) for facile binding to heme iron. Thermal equilibration between configurational ensembles cytMet* and cytHis* takes place before photolysis but is slow compared with heme binding processes after photolysis. Folding of cytMet* ensembles is fast, whereas folding of cytHis* ensembles is slow compared with CO recombination at 1 atm (not shown). A small fraction of unfolded configurations, cytU*, lacks geminate positioning of a labile residue and remains unligated at the sixth axial heme site (until CO recombines).
These conformational subpopulations are presumably in dynamic equilibrium and not persistent on time scales detected by low time resolution (>1 ms) measures of structure such as NMR amide protection experiments. We assume, however, that they are constrained from interconverting on time scales below about 10−4 s by the diffusional times characterizing chain motion. This time scale is in at least rough agreement with previous estimates for the rate of intrachain diffusion in cytochrome c under similar conditions. Hagen et al. (41, 42) estimate a mean time of about 40 μs for diffusion of Met-80 to the heme binding site in the denatured protein. From this estimate, they infer that the minimum time required to form the smallest intrachain loop (about six residues) is 10−6 s, which they suggest sets an upper limit on how rapidly a protein can fold. However, the Met-80 diffusion estimate is model dependent, being based on the kinetic model of Jones et al. (28). In the context of the heterogeneous kinetic model proposed here, we interpret it as a rough upper limit on how rapidly conformations with different heme-ligand proximities interconvert. Guo and Thirumalai obtain a model-independent estimate of about 20 μs for the time required to form a 10-residue loop (43). Given the n3/2 dependence on chain length expected for this time (44), this corresponds to a Met-80 to heme diffusion time of about 300 μs, which is consistent with the interconversion time scale proposed here.
The fractional equilibrium populations of cytMet*-CO, cytHis*-CO, and cytU*-CO in Fig. 3 (represented by the equilibrium constants ka/k−a and kb/k−b) were determined by optimizing the fit of the four intermediate spectra to the corresponding spectra of model cytochromes (Fig. 2). The bis-His and Met–His species were modeled by cytochrome b5 and native cytochrome c, respectively (37). The five-coordinate and CO-ligated species were modeled by the corresponding species in cytochrome c3 (45). The fast and slow observed time constants for CO binding were assigned to the first two pathways and to the third pathway in Fig. 3, respectively. (Although this assignment of CO recombination rates gave the best fit in Fig. 2, this choice was arbitrary in that modeling with other permutations of the CO rebinding rate assignments did not qualitatively affect the results.)
This analysis finds fractional equilibrium populations of 0.22, 0.63, and 0.15, for the cytMet*-CO, cytHis*-CO, and cytU*-CO ensembles, respectively. The higher population of His-binding vs. Met-binding conformations is at least qualitatively consistent with that expected from a random polymer model of the denatured state. The relative concentration of all histidines to all methionines is expected to be about 9 to 1 in a “θ solvent” that just balances intrachain interactions, assuming n values of 10, 17, 49, and 64 for His-26, His-33, Met-65, and Met-80, respectively. The lower ratio implied here, about 3:1, is probably in reasonable agreement given the assumptions and ambiguities in applying such a simple model. Besides the ambiguity inherent in defining n in relation to the heme binding site, the solvent (4.6 M GuHCl) clearly does not completely balance intrachain interactions, because the protein folds in the absence of CO. Indeed, many proteins may require a significantly higher denaturant concentration, 6 M GuHCl, for θ solvation (42, 46). The polymer will be more compact, and effective residue concentration will be proportional to n−1 in a poor solvent (40), in which case the His:Met ratio is predicted to be closer to 4:1.
The kinetic analysis shows further that about 20% of the sample acquires native-like Fe–Met ligation at the same time (about a microsecond after photolysis) that the sample folds to about 10% native helical content, as determined by the TRCD results. We find this correlation between the kinetic results from independent structural probes of protein structure to be significant. It suggests strongly that the microsecond folding process takes place within the cytMet* ensemble at about the same time that it acquires native heme ligation. This inference in turn suggests that folding is global within each molecule of this subpopulation, because each gains roughly half of the helical secondary structure content of the native state. (The MCD spectra presented here cannot distinguish between Met-80 and Met-65 heme ligation. Thus, some fraction of the 20% of sample that acquires Fe–Met ligation may be an Fe—Met–65 component of low helical structure content.)
The key result distinguishing the heterogeneous kinetic model from the homogenous model of Jones et al. (28) is the inability of the homogeneous model to qualitatively reproduce a spectral feature in the MCD spectrum of the bis-His model compound, as shown in Fig. 2C. The microscopic rate constants used in obtaining the multiple-equilibria model results shown in Fig. 2 were fixed by the observed time constants and the respective equilibrium constants for Met and His residue binding, KM and KH, shown in Scheme S1.
Scheme 1.

Homogeneous multiple-equilibria model for heme ligation events after photolysis of cytochrome c-CO.
The equilibrium constants in Scheme S1, not being known a priori, were determined by optimizing the fit of the intermediate spectra to the model spectra. Unlike the heterogeneous mechanism, the multiple-equilibria mechanism is unable to produce a calculated spectrum of the cyt[His–Fe–His]* intermediate that contains all of the qualitative features of the bis-His model spectrum, producing instead a spectrum missing the characteristic negative lobe near 415 nm (38, 45). This distinction between the models is robust in that extensive modification of the multiple-equilibria model was unable to restore the missing feature to the calculated spectrum. Variation of the K values produced a best fit with values significantly larger than those used by Jones et al. Nevertheless, the obligatory replacement of Met by His in this model distorts the spectrum calculated for the Fe–His intermediate. Other variations of the model, such as replacing the single CO recombination time constant used by Jones et al. with permutations of the two observed time constants used in the heterogeneous model produced no qualitative improvement. Neither did including a 1-ms folding step and 1-s back reaction postulated by Jones et al. In contrast, small variations of the heterogeneous model, e.g., permuting CO rate constants, did not compromise the qualitative fit of the 415-nm negative lobe.
The funnel landscape model provides a natural framework for explaining the kinetic heterogeneity in protein folding reported here. Photodissociation of the Fe–CO bond relaxes constraints on protein tertiary structure near the heme, permitting thermal diffusion of unfolded conformations toward near-native conformations. The energy landscape after phototransformation (Fig. 4) comprises a narrow folding funnel centered around the native configuration and a broad shallow basin corresponding to heme ligation by nonnative residues, principally His-33. Although some of the conformational ensembles (cytU*) diffusing on this landscape apparently remain on flat regions of the surface and escape ligation at the vacant axial heme binding site before CO recombines, most diffuse into the free energy minima. Most of these (cytHis*) are captured by biased diffusion into the broad nonnative ligation trap, giving rise to the observed 50-μs time constant. A smaller fraction (cytMet*), however, diffuse directly down the funnel with a rate constant of about 1⋅106 s−1, which coincides with the estimate of Hagen et al. for the fastest folding rate possible in cytochrome c (41, 42). The ligation-trapped ensembles eventually escape and fold with ≈100 millisecond time constants that are limited by the nonnative His off-rate (101−102 s−1), as observed in denaturant dilution studies of oxidized protein (31) and photoreduction (E. C., unpublished data; ref. 27) studies.
Figure 4.

Schematic diagram of the internal free energy (E) landscape for folding after cytochrome c-CO photolysis, cyt*-CO →hν cyt* + CO, as a function of protein configurational coordinates (CC). A folding funnel (N) and an overlapping local minimum (T), corresponding to the histidine-ligation trap, together form a surface topologically similar to the “moat” landscape of Dill and Chan (6) describing heterogeneous (fast and slow) folding kinetics.
The kinetic model presented simplifies what is expected to be a very complex system. Kinetic events at the heme probably involve nonnative coordination by at least two different residues, His-33 and His-26, and perhaps a third, Met-65, as well (28). Thus it is likely that the landscape in Fig. 4 contains several local minima corresponding to the several nonnative ligands known to bind to heme. Furthermore, the kinetic modeling indicates that about 20% of photolyzed protein molecules populate the natively coordinated intermediate seen at about 1−10 μs, whereas the far-UV CD indicates that only about half this percentage of native secondary structure has formed at that time. This observation suggests either that the natively coordinated intermediate has folded only about halfway toward the native secondary structure (we neglect in these estimates the relatively small effect of the denaturant in the absence of CO) or that binding by Met-65 contributes as much as half to the total population of the “natively” coordinated microsecond intermediate. It is thus possible that further folding progress could be observed in this component under conditions where CO recombination is sufficiently delayed.
We consider briefly the presence of nonnative proline isomers as another possible source of early-time kinetic heterogeneity, because proline isomerization is known to be responsible for heterogeneity in the slow folding kinetics of cytochrome c (47). The protein contains prolines at sites 30, 44, 71, and 76. All four are trans in the native structure, whereas the cis content of a given proline is expected to be about 10–30% in the disordered polypeptide (22). Isomer equilibration requires seconds to minutes, depending on the nature of the adjacent residue. Although it is conceivable that proline isomerization may influence heme-residue binding in the unfolded protein, there is to our knowledge no precedent for such an effect, whereas several lines of evidence argue against this possibility. First, structural studies of folded proteins tend to find the effect of proline isomerization to be very localized (48, 49). Second, the lack of stable secondary or tertiary structure in the unfolded protein suggests that such a relatively subtle stereochemical effect will be “washed out” when averaged over disordered chain conformations. Third, the observation that native Met-80 ligation is completely restored before proline isomerization is complete in the folding reactions of both ferri- and ferro-cytochrome c also tends to argue against this idea. Fourth, if Pro-76 influenced Met-80 binding, one would expect the small nonnative cis isomer population present in the denatured protein to give rise to a small Met-binding-impaired component (assuming that the native form has optimum stability) rather than the fast-binding component observed. Finally, perhaps the most direct evidence comes from competitive heme-binding experiments showing that different histidines in the denatured cytochrome c chain bind to heme with effective concentrations that are consistent with simple random polymer statistics (50). In other words, no structural effect such as that of proline isomerization is found to distinguish the heme binding affinities of different histidines. Although these observations do not rule out the possibility that prolyl peptide bond stereochemistry affects differentially the heme binding rates of histidine and methionine residues at least several residues distant on the chain, together they suggest strongly that such effects are probably too small to prevent completely one residue or the other from binding to heme in the denatured protein and so give rise to the heterogeneity observed here. More definitive evidence may be obtained from “double-pulse” (rapid unfolding followed by refolding) experiments or from studies of proline-replaced mutants.
Recent ultrarapid mixing folding studies (51) are beginning to bridge the gap between the time scales available to stopped-flow studies with millisecond dead times and the ultrafast folding time scale examined in the present study. Trp fluorescence studies of oxidized cytochrome c with denaturant dilution mixing times of about 100 μs show the prompt (<50 μs) formation of a compact nascent phase. This phase undergoes heme ligand exchange reactions with time constants that extend over milliseconds, as observed by resonance Raman (52). The millisecond ligand exchange kinetics appear to be homogeneous, implying an upper limit of about 1 ms on the time scale for interconversion of relevant chain conformations. That upper limit and the lower limit found here suggest that the conformational interconversion time lies within the range 10−4−10−3 s.
It has been suggested that the nascent phase observed in ultrarapid mixing studies of cytochrome c (51) and ribonuclease A (53) simply reflects the collapse expected of a random polypeptide on denaturant dilution. Because such a collapse is expected to be a general feature of denaturant dilution experiments even in the absence of folding, this has called into question the general significance of the submillisecond “burst” phase frequently seen in stopped-flow Trp fluorescence studies of protein folding, particularly with regard to the inference of ultrafast folding intermediates (54). However, a recent ultrarapid mixing Trp fluorescence study resolved a ≈50-μs exponential decay process in oxidized cytochrome c that suggests barrier-limited (type I) folding (55). The present finding of an ultrafast folding species in the reduced protein under isotonic conditions suggests further that more structure-sensitive spectroscopies, e.g., far-UV CD, may possibly reveal secondary structure formation in the nascent phase of the oxidized protein as well.
CONCLUSION
We find a small microsecond-folding component in photolyzed samples of guanidine HCl-denatured cytochrome c-CO complex. The correspondence in kinetics and population amplitudes observed by independent structural probes, amide band CD and heme band MCD and absorption, indicates that this component represents global folding in a subpopulation of the sample. We find further that this component represents one of several kinetically independent paths in the early folding events of the reduced protein. Protein configurations with different tertiary structures are distinguished by the proximity of methionine or histidine residues as heme ligands in denatured cytochrome c, allowing the use of TRMCD spectroscopy to monitor conformational ensembles of differing tertiary structure during folding reactions. Although Met and His binding reactions would compete kinetically if both residues have rapid diffusional access to a given heme binding site, our TRMCD results suggest strongly that they proceed as independent parallel processes—processes shown by our photolysis TRCD results and the stopped-flow results of others to correspond to ultrafast and fast folding pathways, respectively. The kinetic decoupling of these folding pathways on the free energy landscape can be explained as a consequence of rates of iso- and endoergic diffusion between ultrafast-folding and hindered-folding configurational subspaces that are too slow to compete with rapid diffusion down the funnel and trap surfaces.
The rough estimate of the conformational interconversion rate constant found here, 103-104 s−1, provides an experimental benchmark for theorists calculating the roughness of the energy landscape that governs diffusion toward folding. The use of submillisecond time-resolved CD and MCD spectroscopies to monitor the evolution of configurational ensembles in cytochrome c highlights the promise of these techniques for exploring the folding landscapes of proteins.
Acknowledgments
We thank Ken Dill, Harry Gray, Tack Kuntz, José Onuchic, Peter Wolynes, and Clare Woodward for helpful comments. This work was supported by National Institutes of Health grants GM-35158 and GM-38549.
ABBREVIATIONS
- TROA
time-resolved optical absorption
- TRCD
time-resolved circular dichroism
- TRMCD
time-resolved magnetic circular dichroism
- GuHCl
guanidine hydrochloride
- MCD
magnetic circular dichroism
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Creighton T E. Biochem J. 1990;270:1–16. doi: 10.1042/bj2700001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim P S, Baldwin R L. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 3.Englander S W, Mayne L. Annu Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 4.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 6.Dill K E, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 7.Chan H S, Dill K E. Proteins: Struct Funct Genet. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 8.Levinthal C. J Chim Phys. 1968;65:44–45. [Google Scholar]
- 9.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazardis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 11.Karplus M, Shakhnovich E. In: Protein Folding. Creighton T, editor. New York: Freeman; 1992. pp. 127–195. [Google Scholar]
- 12.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karplus M, Šali A. Curr Opin Struct Biol. 1995;5:58–73. doi: 10.1016/0959-440x(95)80010-x. [DOI] [PubMed] [Google Scholar]
- 14.Thirumalai D, Woodson S A. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- 15.Shakhnovich E, Gutin A M. Biophys Chem. 1989;34:187–199. doi: 10.1016/0301-4622(89)80058-4. [DOI] [PubMed] [Google Scholar]
- 16.Wolynes P G, Luthey-Schulten Z, Onuchic J N. Chem Biol. 1996;3:425–432. doi: 10.1016/s1074-5521(96)90090-3. [DOI] [PubMed] [Google Scholar]
- 17.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 18.Mines G A, Pascher T, Lee S C, Winkler J R, Gray H B. Chem Biol. 1996;3:491–497. doi: 10.1016/s1074-5521(96)90097-6. [DOI] [PubMed] [Google Scholar]
- 19.Weissman J S. Chem Biol. 1995;2:255–260. doi: 10.1016/1074-5521(95)90044-6. [DOI] [PubMed] [Google Scholar]
- 20.Creighton T E, Goldenberg D P. J Mol Biol. 1984;179:497–526. doi: 10.1016/0022-2836(84)90077-9. [DOI] [PubMed] [Google Scholar]
- 21.Weissman J S, Kim P S. Proc Natl Acad Sci USA. 1992;89:9900–9904. doi: 10.1073/pnas.89.20.9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmid F X, Mayr L M, Mücke M, Schönbrunner E R. Adv Protein Chem. 1993;44:35–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 23.Roder A, Elöve G A, Englander S W. Nature (London) 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jennings P A, Finn B E, Jones B E, Matthews C R. Biochemistry. 1993;32:3783–3789. doi: 10.1021/bi00065a034. [DOI] [PubMed] [Google Scholar]
- 25.Dobson C M, Evans P A, Radford S E. Trends Biochem Sci. 1994;19:31–37. doi: 10.1016/0968-0004(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 26.Gilmanshin R, Williams S, Callender R H, Woodruff W H, Dyer R B. Proc Natl Acad Sci USA. 1997;94:3709–3713. doi: 10.1073/pnas.94.8.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pascher T, Chesick J P, Winkler J R, Gray H B. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 28.Jones C M, Henry E R, Hu Y, Chan C K, Luck S D, Bhuyan A, Roder H, Hofrichter J, Eaton W A. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen E, Goldbeck R A, Kliger D S. Annu Rev Biophys Biomol Struct. 1997;26:327–355. doi: 10.1146/annurev.biophys.26.1.327. [DOI] [PubMed] [Google Scholar]
- 30.Chen E, Wood M J, Fink A L, Kliger D S. Biochemistry. 1998;37:5589–5598. doi: 10.1021/bi972369f. [DOI] [PubMed] [Google Scholar]
- 31.Colón W, Wakem L P, Sherman F, Roder H. Biochemistry. 1997;36:12535–12541. doi: 10.1021/bi971697c. [DOI] [PubMed] [Google Scholar]
- 32.Goldbeck R A, Kim-Shapiro D B, Kliger D S. Annu Rev Phys Chem. 1997;48:453–479. doi: 10.1146/annurev.physchem.48.1.453. [DOI] [PubMed] [Google Scholar]
- 33.Esquerra R M. Ph.D. thesis. Santa Cruz: Univ. of California; 1997. [Google Scholar]
- 34.Henry E R, Hofrichter J. Methods Enzymol. 1992;210:129–192. [Google Scholar]
- 35.Goldbeck R A, Kliger D S. Methods Enzymol. 1993;226:147–177. doi: 10.1016/0076-6879(93)26009-x. [DOI] [PubMed] [Google Scholar]
- 36.Chan C-K, Hofrichter J, Eaton W A. Science. 1996;274:628–629. doi: 10.1126/science.274.5287.628. [DOI] [PubMed] [Google Scholar]
- 37.Vickery L, Nozawa T, Sauer K. J Am Chem Soc. 1976;98:351–357. doi: 10.1021/ja00418a006. [DOI] [PubMed] [Google Scholar]
- 38.Vickery L, Salmon A, Sauer K. Biochim Biophys Acta. 1975;386:87–98. doi: 10.1016/0005-2795(75)90249-4. [DOI] [PubMed] [Google Scholar]
- 39.Treu J I, Hopfield J J. J Chem Phys. 1975;63:613–623. [Google Scholar]
- 40.Flory P J. Statistical Mechanics of Chain Molecules. New York: Wiley; 1969. [Google Scholar]
- 41.Hagen S J, Hofrichter J, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagen S J, Hofrichter J, Eaton W A. J Phys Chem B. 1997;101:2352–2365. [Google Scholar]
- 43.Guo Z Y, Thirumalai D. Biopolymers. 1995;36:83–102. [Google Scholar]
- 44.Szabo A, Schulten K, Schulten Z. J Chem Phys. 1980;72:4350–4357. [Google Scholar]
- 45.O’Connor D B, Goldbeck R A, Hazzard J H, Kliger D S, Cusanovich M A. Biophys J. 1993;65:1718–1726. doi: 10.1016/S0006-3495(93)81226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Damaschun, G., Gast, K., Müller-Frohne, M. & Zirwer, D. Biopolymers, in press.
- 47.Wood L C, White T B, Ramdas L, Nall B T. Biochemistry. 1988;27:8562–8568. doi: 10.1021/bi00423a009. [DOI] [PubMed] [Google Scholar]
- 48.Amodeo P, Morelli M A, Castiglione Motta A. Biochemistry. 1994;33:10754–10762. doi: 10.1021/bi00201a024. [DOI] [PubMed] [Google Scholar]
- 49.Svensson L A, Thulin E, Forsen S. J Mol Biol. 1992;223:601–606. doi: 10.1016/0022-2836(92)90976-q. [DOI] [PubMed] [Google Scholar]
- 50.Muthukrishnan K, Nall B T. Biochemistry. 1991;30:4706–4710. doi: 10.1021/bi00233a010. [DOI] [PubMed] [Google Scholar]
- 51.Chan C-K, Hu Y, Takahashi S, Rousseau D L, Eaton W A, Hofrichter J. Proc Natl Acad Sci USA. 1997;94:1779–1784. doi: 10.1073/pnas.94.5.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeh S-R, Takahashi S, Fan B, Rousseau D L. Nat Struct Biol. 1997;4:51–56. doi: 10.1038/nsb0197-51. [DOI] [PubMed] [Google Scholar]
- 53.Qi P X, Sosnick T R, Englander S W. Nat Struct Biol. 1998;5:882–884. doi: 10.1038/2321. [DOI] [PubMed] [Google Scholar]
- 54.Jennings P A. Nat Struct Biol. 1998;5:846–848. doi: 10.1038/2286. [DOI] [PubMed] [Google Scholar]
- 55.Shastry M C R, Roder H. Nat Struct Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]