

Abstract
In this work we describe protocols for the generation of gene deletions and gene replacements using a temperature sensitive plasmid in Escherichia coli O157:H7. This technology requires flanking DNA to be cloned into a temperature sensitive vector but the resulting clone allows great flexibility for further modification of the target sequence. It is therefore highly suited to the study of genes in which several rounds of changes are envisaged. A number of examples are used to illustrate the flexibility of the system which has been used to create novel gene replacements including fusions for protein localisation work and reporters for transcriptional analyses. In this paper we describe protocols which can be used with a high degree of success when applied to E. coli O157. The deletion and replacement of the LEE4 operon of E. coli O157 is detailed to show the advantages and limitations of the technology.
Keywords: Gene Deletion, Escherichia coli O157:H7
Introduction
The ability to generate directed gene deletions and gene replacements is a central technique used to demonstrate gene function and fulfil Molecular Koch’s postulates in any study (1). The past two decades have seen the development of a variety of molecular techniques to achieve such chromosomal modifications in E. coli K-12, including F’ homogenotization, the formation and resolution of ColE1 plasmid cointegrates in a polA1 background, transformation and mating via an Hfr cross, and the use of temperature sensitive plasmids with selection for homologous recombination (2, 3). More recently, the phage λ red recombinase has been successfully applied to allow rapid, one step inactivation of genes (4). However, these techniques can suffer from the requirement of specific genetic backgrounds (often mutants themselves), and are optimized for efficient use in E. coli K-12 laboratory strains. The application of these techniques to “wild-type” strains including pathotypes of E. coli is, therefore, often problematic. Furthermore, using temperature sensitive plasmids provides a number of key advantages compared with other methods. Firstly, although lambda red is probably the most rapid system for inactivation of genes it does suffer from the fact that transient expression of the recombinase system enhances the frequency of unwanted genetic rearrangements. Secondly, the lambda red approach is also highly suited to creation of gene deletions, delivery of gene replacements, as we describe for the temperature sensitive based system, is less straightforward. Additionally, as the temperature sensitive allelic exchange system does not rely on any conjugation step, for example when using lambda Pir-dependent iroR6K plasmids, strains that produce bacteriocins or capsule can also be successfully modified.
In the case of E. coli O157, the complete genome sequencing of two strains has revealed an additional 1.4MB of DNA when compared with the E. coli K-12 genome (5). These sequences include numerous putative adhesin clusters, the complete LEE pathogenicity island, shiga-like toxins encoded on prophages and other putative virulence factors (6). Genetic manipulation of E. coli O157 to selectively disrupt these factors is therefore highly desirable. Two approaches have been widely adopted to achieve this goal: a modified lambda red protocol and the use of temperature sensitive plasmids and selection for allelic exchange (7, 8). Here we describe a modified protocol for use with temperature sensitive plasmids. This protocol reliably yields allelic exchange constructs, works well to replace large DNA fragments (we have deleted/modified up to 7KB), and gives excellent flexibility for further modifications. This flexibility is demonstrated by our use of the system to: 1) knockout a specific operon and then complement the knockout by allelic exchange back at the site of deletion. 2) Engineer whole gene or sequence deletions. 3) Introduce marker genes for single copy reporter fusions. 4) Introduce specific tags to track or visualise the resultant recombinant proteins.
Outline of Allelic Exchange Methodology
In this paper we describe a modified allelic exchange method that allows the exchange of genetic information into a wild-type E. coli strain background. Firstly an intermediate strain is constructed in which the wild-type chromosomal sequence is replaced by a DNA cassette. This sacB-kan cassette contains the sacB gene from Bacillus subtilis and the neomycin (kanamycin) gene from Tn5. The method has two distinct rounds of allelic exchange. The sacB-kan cassette is cloned into a temperature sensitive chloramphenicol resistant plasmid in between homologous flanking regions of DNA. This is used in the first round of allelic exchange to create an intermediate strain containing the cassette on the chromosome in replace of wild-type sequence (Fig. 1). The selection for plasmid integration occurs at 42°C in the presence of chloramphenicol while plasmid excision occurs at 28°C in the presence of kanamycin. In the second round of allelic exchange, for example to create a complete deletion, the temperature sensitive plasmid containing only the homologues flanking regions is used (Fig. 2). Alternatively DNA cloned in between the flanking regions can be exchanged onto the chromosome e.g. to complement the mutant or to introduce different alleles. During this allelic exchange the plasmid is transformed into the intermediate strain. Growth at 42°C in chloramphenicol selects for plasmid integration, whilst growth at 28°C in the presence of sucrose selects for bacteria in which the inserted cassette has been exchanged for the target sequence and then the plasmid lost through lack of selective pressure. The selective pressure results from the action of the sacB gene that encodes levansucrase, an enzyme that cleaves sucrose resulting in toxic levels of levans in many E. coli strains.
Fig. 1. (Adapted from Blomfield et al. 1991).
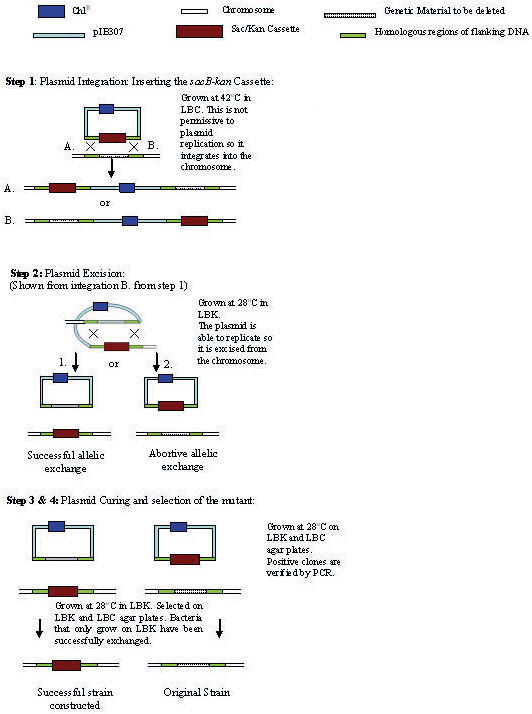
The transfer of the sacB-kan cassette into the chromosome requires two recombination events leading to plasmid integration followed by plasmid excision. Step 1: plasmid integrates into the wild-type strain at the non permissive temperature for plasmid replication (42°C). Steps 2 and 3: plasmid integrates are grown at 28°C in the presence of kanamycin to enrich for bacteria that have excised and cured the plasmid, leaving the cassette on the chromosome. Step 4: Growth on media containing LBC or LBK. Bacteria that can only grow on LBK are successful constructs of the intermediate strain.
Fig. 2. (Adapted from Blomfield et al. 1991).
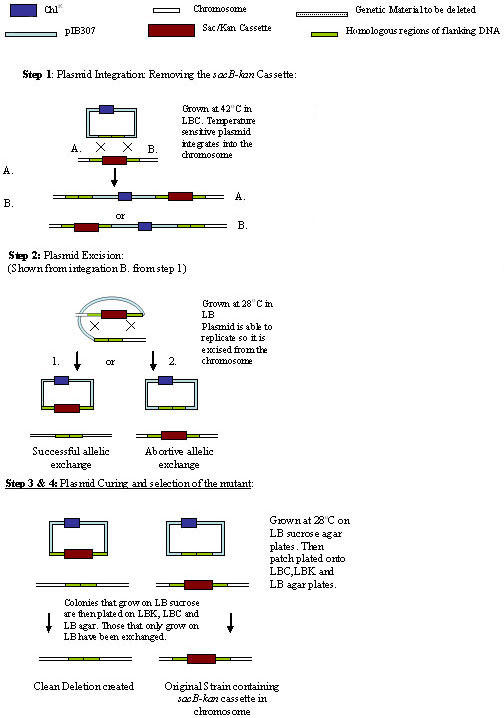
Step 1: Plasmid integration in the intermediate strain is selected at the non permissive temperature for plasmid replication (42°C). Steps 2 and 3: Plasmid integrates are grown at 28°C in the absence of antibiotics to enrich for bacteria that have excised (Step 2) and later cured (Step 3) the integrated plasmid. Step 4: Growth on media containing sucrose selects for successful strain constructs.
Materials and Methods
The allelic exchange method has been used for a number of studies in which defined deletions or gene replacements were made. In this paper, the deletion and complementation of the LEE4 operon of Escherichia coli O157 is used as a specific example to illustrate the technique. A number of further examples are used to illustrate how the technology can be used in other applications.
Creation of the LEE4 deletion and complemented strains
The allelic exchange method was used to delete and then repair the entire LEE4 operon from E. coli O157:H7 (ZAP198), generating strains ZAP984 and ZAP985, respectively. A step by step protocol for the allelic exchange procedure is listed under the protocols section of this work. These methods were used to create ZAP984 (ΔLEE4), using plasmid pHY10 and the complemented strain, ZAP985, using plasmid pAJR162. The detailed construction of these plasmids and strains including oligonucleotide sequences are covered in Naylor et al., 2005 (9). In this work, we aim to provide an overview of the steps involved in the cloning, the allelic exchange methodology and our experience with the technology.
Results and Discussion
Factors affecting co-integrate formation
Two key factors play a role in determining the frequency of co-integrate formation: 1) degree of DNA homology and 2) size of the flanking regions. We have observed a dramatic reduction in efficiency of primary integrate formation with even a few base pair mismatched between flanking region DNA and target region on the chromosome. This is a drawback as sometimes it results in the requirement of cloning specific flanking regions for each strain being studied. For example, we have used an allelic exchange system at the lac locus in several wild-type O157:H7 strains that created a number of promoter-lacZ fusions (8). However, we observed extremely low frequencies of allelic exchange when plasmids were used with any strain other than the one from which the flanking regions were cloned.
The key reagent we use for the knockout strategy is the plasmid pIB307. The replicon from this plasmid is derived from the large antibiotic resistance plasmid R6-5 (10). This replicon and a tetracycline resistance gene were manipulated to form a smaller plasmid termed pSC101. pSC101 has undergone in vitro mutagenesis with hydroxylamine, creating distinct mutants that varied in their temperature dependent replication (11). These included pH01, a pSC101 replicon mutant that was unable to replicate at 42°C but could replicate normally at 28°C. The replicon from pH01 was removed and ligated to the chloramphenicol resistance gene from pBR325. The resulting plasmid, pMAK705, was modified by the addition of the lacZ gene and its promoter (to facilitate screening) and the polylinker M13mp19 as a multiple cloning site (12). At a later date the majority of the lac sequences were removed to decrease the plasmid’s size and homology with chromosomal DNA, thereby creating pIB307 (13).
For flanking regions we routinely use between 600bp and 1000bp of DNA, and find it is important to have similar sizes of left and right hand flanking regions to maximise the efficiency of exchange. Grossly differing flanking regions will favour recombination at one flanking region thereby reducing frequency of the double crossover event that is required for successful allelic exchange. The deletion of LEE4 used a left hand flanking region of 958 bp and a right hand flanking region of 610 bp. Smaller flanking regions (200-300bp) can work but will reduce the efficiency of exchange. Each flanking region for the LEE4 knockout was PCR amplified and cloned into the temperature sensitive vector pIB307 creating pHY3. A sacB-kan cassette was cloned between the flanking regions at the BamHI restriction site to create plasmid pHY10 (8). Again, the requirement of cloning three separate DNA fragments in the creation of the allelic exchange vector is a disadvantage compared with the lambda red approach. To allow subsequent ‘repair’ of any deletion, the LEE4 operon was amplified by long-range PCR and cloned into pIB307 to create construct pAJR162.
An alternative method is to amplify the whole region including the flanking regions and the gene(s) to be deleted using long-range PCR. This can be cloned into the pIB307 vector and restricted by endonucleases at naturally occurring restriction sites to replace the gene(s) to be deleted with the sacB-kan cassette. This plasmid can then be used in the allelic exchange process.
Optimising conditions for generation of large deletions
For large deletions of several kilobases or problematic deletions we have found that increasing the temperature from 42°C to 44°will aid the selection of primary integrates. Furthermore, we also find it useful to plate out dilutions of the primary integrates on LBC plates at 44°C, enabling single colonies to be analysed for the primary integration event by PCR. This “primary positive” colony can then be cultured at the lower, permissive temperature of 28°C to allow the generation of resolution products. This approach is particularly useful if any construct results in low levels of primary integrates at 42°C, often indicated by a subsequent failure to culture at 28°C.
Location of the sacB gene
The sacB gene offers a powerful tool for the selection of allelic exchange events that do not occur frequently. The stage at which the sacB gene carries out this function in the allelic exchange strategy can vary depending on its location on the exchange plasmid. For the creation of the LEE4 deletion strain the exchange plasmid contained the sacB gene as part of a cassette that was situated in between the homologous flanking regions. For the complementation of LEE4 the sacB gene was used to select for bacteria that had lost the sacB-kan cassette from the chromosome after it was used to replace the genetic material, i.e. on the second round of allelic exchange. If the sacB gene is located outwith the homologous flanking regions then it can be used as a negative selective marker for the loss of the plasmid during the first round of allelic exchange. The colonies that do not contain the sacB gene successfully grow in the presence of sucrose and can then be screened for the correct resistance profiles (if any) and verified by PCR. In one study the sacB gene was situated on the allelic exchange plasmid outside the flanking regions. In between the flanking regions was a cassette that replaced the genetic material of interest for a genetic element containing lacZ, the aph gene (encoding resistance to kanamycin) and the lac promoter. This cassette was flanked by FRT sites, so it could be excised from the chromosome by the Flp recombinase provided in trans, creating a clean deletion (14).
Exchange frequencies
During the first round of allelic exchange when the sacB-kan cassette is inserted into the chromosome, we have found that >80% of colonies plated on LBC after 48 hours selection are primary integrants suitable for resolution (ascertained by PCR). Following culture at 28°C, the success of the allelic exchange can vary widely, from 5-90%. Therefore, we typically replica plate about 100 colonies and expect to find a successful exchange clone. The removal of the sacB-kan cassette during the second round of exchange has a higher frequency of success due to the negative selective screening offered by the sacB gene.
Construct validation
Following the allelic exchange process and, assuming a clone has been obtained with the correct antibiotic resistance profile; two key checks should be carried out on any construct. Firstly, the loss of the plasmid is verified using PCR primers designed to amplify the pIB plasmid (Table 1). Secondly, the region that has been exchanged is PCR amplified (usually in 2 or more sections) including DNA that is outside the flanking region, i.e. was unmodified during the exchange process. This shows that the allelic exchange is indeed at the target site and that the cloned DNA does not contain any sequence errors.
Table 1.
Strains and oligonucleotide primers used in the study.
| Strains | Description |
| ZAP198 | E. coli O157:H7 (Walla3) VT1-VT2- |
| ZAP984 | ZAP198 Δ LEE4 |
| ZAP985 | ZAP784 LEE4 complement |
| Primers | Primer Sequence |
| PIB sp.3 | AGACAAATGGATCTCGTAAGCG |
| PIB sp.5 | GCTGTAACAAGTTGTCTCAGGTGT |
| Plasmids | Description |
| pIB307 | pMAK705 based plasmid for allelic exchange. Temperature sensitive replicon. |
| pAJR162 | pIB307 vector with cloned LEE4 |
Application of the Method
Examination of EspD secretion phenotype in wild-type, deletion and complemented strains
The ability of strains to secrete EspD protein in the wild-type, and LEE4 deletion strain (ZAP984) and the complement (ZAP985) was confirmed by Western analysis. As expected, deletion of LEE4, the operon that encodes EspD, leads to a loss of secretion phenotype. Complementation of the deletion mutant by allelic exchange using the LEE4 operon resulted in restoration of the secretion phenotype. Clearly, complementation using plasmids is far easier than the approach described. However, the approach has a number of limitations, including plasmid stability and copy number. We subsequently used the LEE4 deletion strain and complement to assess the importance of this operon during colonization of the terminal rectum of cattle (9). The LEE4 complement strain was capable of adhering to bovine epithelium in vitro and colonising the terminal rectum of cattle, indicating that the type III secretion system plays a key role in the animal host. In such an animal model, plasmid selection using antibiotics is not always possible and many studies therefore avoid performing any form of complementation. We consider that single-copy chromosomal complementation is more desirable, as it can replace the region of interest into its original site thereby fulfilling molecular Koch’s postulates (1).
Further applications
We are currently using the described allelic exchange system to deliver a number of hybrid protein fusions into the structural components of the type three secretion system. This approach includes delivery of an EscU::eGFP fusion (EscU being a type three secretion apparatus protein) into the chromosome and subsequent hybrid protein localisation using fluorescence microscopy and Western analyses. Ongoing work is also replacing the eGFP protein with brighter variants such as GFP+ and alternative tags such as haemagglutinin (HA) to determine the most appropriate detection system in terms of sensitivity and stability. Again, the flexibility of the system allows easy replacement of the sacB-kan cassette with any construct cloned between the flanking region DNA.
We have made extensive use of the described system to create chromosomal lacZ fusions to fimbrial genes, type III secretion genes and regulators (15-17). In these studies we observed that once the sacB-kan cassette is integrated into the chromosome, it is straightforward to introduce single copy fusions at this target site
Potential problems areas
In our experience, there are two inherent problems with the method. Firstly, the pSC101 replicon based plasmid vector can be susceptible to loss of temperature sensitivity. Care must be taken to ensure that all initial cloning steps, transformation recoveries and cultures are carried out at 30°C or less. Loss of temperature sensitivity will result in the inability to cure the strain of the plasmid. We have even encountered this problem following particularly hot summer days when the temperature of the laboratory could reach >28°C. Secondly, the copy-number of the plasmid is approximately 6 to 10 per cell. For certain genes it is possible that this could result in gene dosage problems. This may explain our observation when we attempted to delete a DNA methylase gene but found the initial transformants cultured very slowly in LBC broth and did not produce primary integrates (data not shown). With regards to which strains of E. coli the technique can be applied to, we have successfully manipulated several E. coli O157 strains, enteropathogenic E. coli (EPEC), uropathogenic E. coli (UPEC) and K-12 strains. The only strain-specific factor that may limit the application of the technique is if the strain carries a sucrose utilisation system that has been described in certain isolates of E. coli (18). Clearly the presence of such loci would negate the selective capability of the sacB gene used in the allelic exchange protocol.
It is also important to appreciate that unintended secondary mutations can occur when using suicide plasmids to create a deletion (19). Before a phenotypic change is attributed to the deleted gene the strain should be checked. PFGE has been shown to be a useful tool for detecting these unwanted changes and Southern blotting is also advisable to show that no rearrangements have occurred proximal to the site of the targeted modification. Alternatively, reverse allelic exchange can restore the wild-type allele to the genome; if the phenotype is restored then the changes observed can be attributed to the deletion.
Summary
Allelic exchange is an invaluable tool for creating defined deletions of chromosomal DNA sequences. In this paper we describe protocols that ensure a high degree of success when using temperature sensitive plasmids for generating deletions in Escherichia coli O157:H7. Our experience in creating defined knockouts and complements at several loci is discussed as are potential problem areas. Further applications of the technology are covered including the creation of chromosomal gene fusions to markers such as green fluorescent protein and haemagglutinin.
Figures
Acknowledgments
J.R.E. is supported by a BBSRC funded studentship. D.L.G. and A.J.R. are supported by funding from the Department of Environment, Food and Rural Affairs (DEFRA) VTRI Fellowship. The authors have no conflicts of interest to declare related to this publication.
Appendix
Protocols
The exchange process relies on a series of sub-culturing steps. Each time 100 μl of the culture is used to inoculate 100 ml of fresh media. The old culture is discarded and the new inoculated media is incubated for 8 or 16 hours.
Inserting the sacB-kan cassette into the chromosome:
Day One:
Transform the exchange plasmid into the appropriate strain
Plate 250 μl of the recovered transformation onto LB (Luria-Bertani) agar plates containing chloramphenicol (25 μg/ml)
Recover at 28°C in a static incubator for ~48 hours
Day Three:
Inoculate 100ml of LBC broth (Luria-Bertani broth containing 25 μg/ml of chloramphenicol) with ~10 colonies from the recovered transformation plate
Incubate at 42°C and 200 RPM in a shaking incubator for ~16 hours
Day Four:
Sub the culture into 100 ml LBC broth, incubate for ~8 hrs at 42°C and 200 RPM
After 8 hrs repeat the sub-culturing step into LBC and incubate for ~16 hrs at 42°C and 200 RPM
Day Five:
Duplicate the sub-culturing step into LBC broth and incubate for ~8 hrs at 42°C and 200 RPM
After 8 hrs sub the culture into 100 ml of LBK (Luria-Bertani broth containing 25 μg/ml of kanamyacin) and incubate for ~16 hrs at 28°C and 200 RPM
Before discarding the culture used for the LBK sub create serial dilutions and plate 250 μof the 10-4 and 10-5 dilutions onto LBC agar plates. Incubate at 28°C in the static incubator for 16 hours. These can be used to screen for primary integrants if later steps in the allelic exchange process fails
Day Six:
Sub the culture into 100 ml of the LBK broth and incubate for ~8 hrs at 28°C and 200 RPM
Repeat the sub-culturing step into LBK broth and incubate for ~16 hrs at 28°C and 200 RPM
Day Seven:
Repeat the sub-culturing step into LBK broth and incubate for ~8 hrs at 28°C and 200 RPM
After 8 hrs create serial dilutions of the culture and plate out 250 μl of the 10-3, 10-4, 10-5 and 10-6 dilutions onto LBK agar plates and incubate for ~16 hrs in a static incubator at 28°C
Day Eight:
Use the colonies from the serial dilution LBK agar plates to create patch plates. Each colony screened should be patched on an LBC agar plate first followed by an LBK plate. Only those colonies that grow on LBK and not LBC will be correct. On average 100 – 200 colonies are screened.
The colonies that can only grow on the LBK patch plate are further checked by PCR. They can be screened by using pIB specific primers. Primers that sit inside the deleted genetic material can also be used. The correct strain should only grow on kanamyacin.
Removing the sacB-kan cassette from the chromosome, leaving a clean deletion:
Day 1:
Transform the exchange plasmid (containing the flanking regions and no sacB-kan cassette) into the intermediate strain with the sacB-kan cassette in the chromosome
Plate 250 μl of the recovered transformation onto LB (Luria-Bertani) agar plates containing chloramphenicol (25 μ/ml).
Recover at 28°C in a static incubator for ~48 hours
From this the steps are identical to inserting the sacB-kan cassette into the chromosome, up until day four, where the subs go from LBC to LB (not LBK). This is grown and sub-cultured (into LB) in the same way until day seven when the dilutions are plated onto LB agar plates containing 6% sucrose and no NaCl. This is a selective media where any bacteria still containing the sacB-kan cassette will not be able to grow; the colonies that do grow are patched onto LBK, LBC and LB (or replica plated). Only those colonies that grow on LB and not LBK or LBC are correct and can be verified by PCR.
References
- Falkow S. Molecular Koch's postulates applied to bacterial pathogenicity - a personal recollection 15 years later. Nat Rev Microbiol. 1994;2:67–72. doi: 10.1038/nrmicro799. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. New York: Cold Spring Harbour Laboratory Press; 1972.
- Parker B, Marinus MG. A simple and rapid method to obtain substitution mutations in Escherichia coli: isolation of a dam deletion/insertion mutation. Gene. 1988;73:531–535. doi: 10.1016/0378-1119(88)90517-3. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna NT et al . Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- Spears KJ, Roe AJ, Gally DL. A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol Letts. 2005;255:187–202. doi: 10.1111/j.1574-6968.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- Campellone KG, Robbins D, Leong JM. EspF(U) is a translocated EHEC effector that interacts with Tir and N-WASP and promotes nck-independent actin assembly. Dev Cell. 2004;7:217–228. doi: 10.1016/j.devcel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Roe AJ et al . Heterogeneous surface expression of EspA translocon filaments by Escherichia coli O157:H7 is controlled at the posttranscriptional level. Infect Immun. 2003;71:5900–5909. doi: 10.1128/IAI.71.10.5900-5909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor SW et al . Escherichia coli O157:H7 forms attaching and effacing lesions at the terminal rectum of cattle and colonization requires the LEE4 operon. Microbiol. 2005;151:2773–2781. doi: 10.1099/mic.0.28060-0. [DOI] [PubMed] [Google Scholar]
- Cohen SN, Chang ACY. Revised Interpretation of the Origin of the pSC101 Plasmid. J Bacteriol. 1997;132:734–737. doi: 10.1128/jb.132.2.734-737.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Gotoh TSM, Sekiguchi M. Mutations to Temperature Sensitivity in R Plasmid pSC101. J Bacteriol. 1977;131:405–412. doi: 10.1128/jb.131.2.405-412.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CM et al . New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989;171:4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomfield IC et al . Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol. 1991;5:1447–1457. doi: 10.1111/j.1365-2958.1991.tb00791.x. [DOI] [PubMed] [Google Scholar]
- Merlin C et al . Tools for characterization of Escherichia coli genes of unknown function. J Bacteriol. 2002;184:4573–4581. doi: 10.1128/JB.184.16.4573-4581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low AS et al . Analysis of fimbrial gene clusters and their expression in enterohaemorrhagic Escherichia coli O157:H7. Env Microbiol. 2006;8:1033–1047. doi: 10.1111/j.1462-2920.2006.00995.x. [DOI] [PubMed] [Google Scholar]
- Roe AJ et al . Co-ordinate single-cell expression of LEE4- and LEE5-encoded proteins of Escherichia coli O157:H7. Mol Microbiol. 2004;54:337–352. doi: 10.1111/j.1365-2958.2004.04277.x. [DOI] [PubMed] [Google Scholar]
- Porter ME et al . The LEE1 promoters from both enteropathogenic and enterohemorrhagic Escherichia coli can be activated by PerC-like proteins from either organism. J Bacteriol. 2005;187:458–472. doi: 10.1128/JB.187.2.458-472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunekawa H et al . Acquisition of a sucrose utilization system in Escherichia coli K-12 derivatives and its application to industry. Appl Environ Microbiol. 1992;58:2081–2088. doi: 10.1128/aem.58.6.2081-2088.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR et al . High-Frequency Secondary Mutations after Suicide-Driven Allelic Exchange Mutagenesis in Extraintestinal Pathogenic Escherichia coli. J Bacteriol. 2003;185:5301–5305. doi: 10.1128/JB.185.17.5301-5305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]