

Abstract
Background:
Non-long terminal repeat (non-LTR) retrotransposons are eukaryotic mobile genetic elements that transpose by reverse transcription of an RNA intermediate. We have performed a systematic search for sequences matching the characteristic reverse transcriptase domain of non-LTR retrotransposons in the sequenced regions of the Drosophila melanogaster genome.
Results:
In addition to previously characterized BS, Doc, F, G, I and Jockey elements, we have identified new non-LTR retrotransposons: Waldo, You and JuanDm. Waldo elements are related to mosquito RTI elements. You to the Drosophila I factor, and JuanDm to mosquito Juan-A and Juan-C. Interestingly, all JuanDm elements are highly homogeneous in sequence, suggesting that they are recent components of the Drosophila genome.
Conclusions:
The genome of D. melanogaster contains at least ten families of non-site-specific non-LTR retrotransposons representing three distinct clades. Many of these families contain potentially active members. Fine evolutionary analyses must await the more accurate sequences that are expected in the next future.
Background
Non-long terminal repeat (non-LTR) retrotransposons, also known as LINEs (long interspersed nuclear elements), make up a very large class of transposable elements that are present in most eukaryotes [1,2]. They transpose by reverse transcription of an RNA intermediate. They possess an open reading frame (ORF) with coding capacities for a protein with endonuclease, reverse transcriptase and sometimes RNase H activities. Many of them contain an additional ORF (ORF1) encoding a protein of an unknown function. Some non-LTR retrotransposons, such as R1 and R2 in arthropods, insert at specific sites in the genome [3,4], whereas the others do not show specific sites of insertion. The cleavage specificity of some elements such as R2 is the result of the activity of an endonuclease showing similarities to restriction enzymes [5,6]. The other elements, whether they are site-specific (like R1) or not (like mammalian L1 elements), have an endonuclease structurally related to apurinic/apyrimidinic (AP) endonucleases [7,8]. Non-LTR retrotransposons can be assigned to 12 clades on the basis of the sequences of their reverse transcriptase domains and appear to be as old as eukaryotes [2,9]. The chronology of the acquisition of the various enzymatic domains suggests that the first non-LTR retrotransposons contained an endonuclease related to restriction enzymes, and that the non-LTR retrotransposons that have a nuclease structurally related to AP endonucleases derived from this ancestral group. The RNase H domain would have been acquired later [2].
Reverse transcription of non-LTR retrotransposons is thought to occur on chromosomal DNA using the cut resulting from their endonuclease activity as a primer. This process is called target-primed reverse transcription (TPRT). The retrotransposition machinery of non-LTR retrotransposons is thought to be used for transposition of SINEs (short interspersed nuclear elements) and might also be at the origin of the formation of processed pseudogenes [10]. The shape of eukaryotic genomes is therefore largely influenced by non-LTR retrotransposons. They are particularly abundant in mammals. Humans contain one major class of non-LTR retrotransposons, the L1 elements, which make up more than 15% of the genome [11]. In contrast, the Drosophila melanogaster genome has more than ten non-LTR retrotransposon families, representing the Jockey, I, R1 and R2 clades [2,9], indicating that several elements have had the chance to spread in this species. Early studies of several families of non-site-specific Drosophila non-LTR retrotransposons have shown that, in many cases, they comprise recently transposed euchromatic elements dispersed on all chromosomal arms, and variously defective elements accumulated in pericentromeric heterochromatin [12,13]. Euchromatic elements can be full size or truncated at the 5' end, presumably as the result of early arrest of reverse transcription. Heterochromatic elements are retrotranspositionally inactive and represent old components of the genome.
Many non-LTR retrotransposons in D. melanogaster were discovered during analysis of spontaneous mutations, as a result of their insertion within a gene. The sequence of most of the euchromatic part of the D. melanogaster genome has recently been reported [14], giving an interesting opportunity for studying all the non-LTR retrotransposons present in this species.
Results
Our aim was to identify all families of non-LTR retrotransposons in the sequenced part of the D. melanogaster genome. For this, we used software based on profile hidden Markov models (HMMs) to find all sequences matching the full-length reverse transcriptase model and containing the conserved motif [FY]XDD (in single-letter amino acid code) [15]. This approach makes it possible to simultaneously identify all potential reverse transcriptase sequences, including LTR elements, non-LTR elements and retroviruses. To distinguish between these classes of retrotransposons, sequences resulting from HMM search were used in BLAST searches against all known retrotransposons, and were assigned by the best match to the three groups. In our analyses, only the non-LTR fraction of the HMM search results was extracted for detailed investigation. Release 1 of the D. melanogaster genome sequence [14] and the Berkeley/European Drosophila Genome Projects (BDGP/EDGP) sequences [16] were analyzed. In both datasets, reverse transcriptase sequences of many known D. melanogaster non-LTR retrotransposons were identified. In addition, some reverse transcriptase sequences with highest similarity to retrotransposons from non-Drosophila species were recognized, providing a basis for identification of new families (see below). The results of these analyses are represented in Figure 1.
Figure 1.
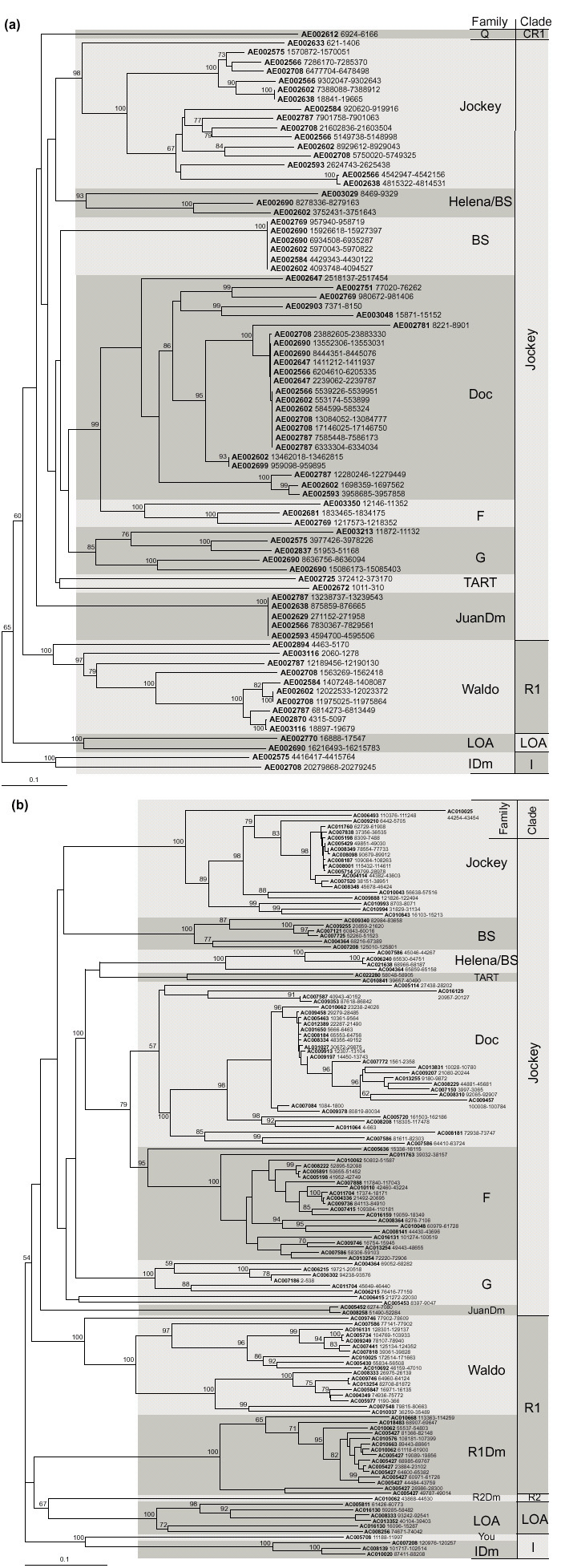
Reverse transcriptase sequences found in the genome of Drosophila melanogaster. Phylogenetic trees of sequences matching the full-length hidden Markov model of reverse transcriptase as defined by HMMER 2.1.1 that were identified in the analysis of (a) Release 1 and (b) BDGP/EDGP sequences. Names on branches represent GenBank accession numbers followed by coordinates in the respective sequence, which confine the HMM match (start coordinate greater than end means reverse complement of a sequence). Distinct families are separated by clades according to [2]. The tree was constructed by the neighbor-joining method as implemented in CLUSTAL W software. Numbers at the nodes represent bootstrap values as percentages out of 500 replicates and are shown only for values greater than 50%. Because of redundancy present in the BDGP/EDGP data set, absolutely identical sequences were substituted by one representative each in building the tree in (b).
Each family of non-LTR retrotransposons identified in this way was further analyzed at the nucleotide level: BLASTN searches were performed, both in the sequences from Release 1 [14] and from BDGP/EDGP [16], to identify all members of the family in the genome, and full-size copies were identified. The BDGP/EDGP sequences appear to be a subset of Release 1 sequences. There is, however, a high error rate in the sequences of Release 1 containing repetitive DNA: comparison of a fraction of both datasets revealed 0.42% point differences in repetitive sequences compared with only 0.0046% point differences in non-repetitive sequences (see [17] and the Celera website [18]). In our analysis, we estimate that non-LTR retrotransposon sequences that could be found in both BDGP/EDGP and Release 1 datasets contain an average of one difference in every 300 base pairs (bp), that is 0.33%. About 40% of these differences are due to insertions and deletions rather than base mismatches, and they are associated with resolution of repeated residues: for example, 5 G should be 6 G, 3 T should be 2 T, 3 C should be 2 C, 8 A should be 7 A, and so on. Consequently, many full-size elements from Release 1 appear not to have coding capacities for complete ORF1 and ORF2 products because of frameshifts. We therefore chose as a representative of each new family identified a full-length element extracted from the BDGP/EDGP database.
The results of our searches are summarized in Table 1. Most of the non-LTR retrotransposon families that we found were already identified and described. These include BS, Doc, F, G, I and Jockey. We have identified three new families: Waldo, JuanDm, and You. They belong to the R1, Jockey and I clades [2], respectively (Figure 2). Not surprisingly, no full-size copy of elements from the R1Dm, R2Dm, TART and HeT-A elements were identified, neither in Release 1 nor in the BDGP/EDGP sequences. Presumably this is because rDNA (target of R1Dm and R2Dm) and telomeric DNA (target of HET-A and TART) have been excluded from the sequencing project. Finally, sequences related to Q, LOA, Bilbo and Helena were found, but no full-size copy was identified for these families.
Table 1.
Summary of non-LTR retrotransposon sequences found in the genome of the D. melanogaster isogenic strain y; cn bw sp
| Number of elements* | Localization† | Full-size elements‡ | 5' truncated§ | D¶ | |||||
| Family | Total | Unfinished | Euchromatic | ? | With TSD | Without TSD | With TSD | Without TSD | >200 |
| BS | 19 | 5 | 15 | 4 | 2 | 0 | 5 | 3 | 0.055 |
| Doc | 128 | 38 | 78 | 50 | 18 | 4 | 20 | 14 | 0.095 |
| F | 94 | 25 | 59 | 35 | 2 | 12 | 7 | 10 | 0.079 |
| G | 37 | 3 | 22 | 15 | 1 | 0 | 2 | 12 | 0.219 |
| I | 48 | 15 | 20 | 28 | 6 | 0 | 6 | 8 | 0.097 |
| Jockey | 86 | 16 | 78 | 8 | 7 | 0 | 40 | 6 | 0.036 |
| JuanDm | 8 | 1 | 8 | 0 | 4 | 0 | 2 | 0 | 0.003 |
| Waldo-A | 82 | 20 | 40 | 42 | 2 | 0 | 10 | 10 | 0.119 |
| Waldo-B | 48 | 4 | 24 | 24 | 4 | 1 | 3 | 17 | 0.137 |
| You | 19 | 3 | 6 | 13 | 3 | 1 | 1 | 2 | 0.116 |
| QDm | 85 sequences matching reverse transcriptase | 0.435 | |||||||
| LOA | 18 sequences matching reverse transcriptase | 0.370 | |||||||
| HelenaDm | 9 sequences matching reverse transcriptase | 0.594 | |||||||
| TART | 11 sequences matching reverse transcriptase | 0.374 | |||||||
| R1Dm | Only partial elements | 0.417 | |||||||
| R2Dm | No hit | ||||||||
| Bilbo | Only short (70-600 bp) regions with average identity of 55% | ||||||||
| HeT-A | Only short (70-600 bp) regions with average identity of 60% | ||||||||
*The total number of elements of each family and the number of elements that were not completely sequenced are indicated. †The localization of elements on chromosomal bands was deduced from the scaffold mapping [14]. Question mark indicates elements whose sequences were found in scaffolds that were not mapped to chromosomal arms. ‡§The number of full size and 5'-truncated elements, surrounded and not surrounded by TSD, are given. ¶The mean genetic distance between elements longer than 200 bp.
Figure 2.
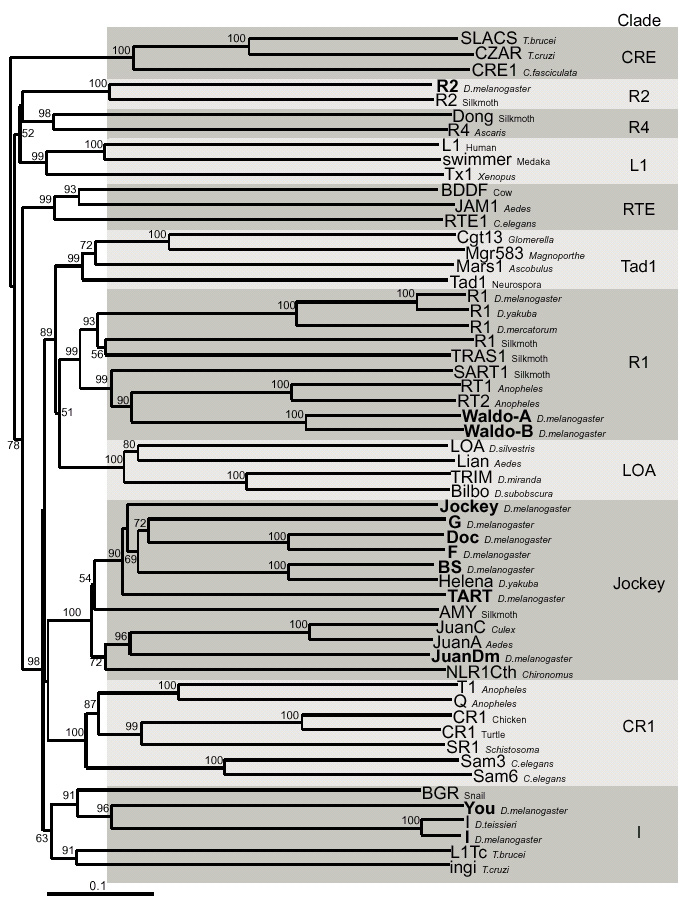
Phylogeny of non-LTR elements based on their reverse transcriptase domain. Drosophila melanogaster non-LTR retrotransposons are shown in bold on the phylogenetic tree based on reverse transcriptase sequences [2]. Sequence alignment was produced by CLUSTAL W software by adding Waldo, JuanDm and You sequences to the alignment DS36752 [2]. Numbers at the nodes represent bootstrap values as percentages out of 500 replicates. The unrooted neighbor-joining tree and bootstrap analysis were inferred as implemented in the MEGA package [57].
Elements of the Jockey clade
The Jockey clade is by far the most represented clade in D. melanogaster. It includes one site-specific element, TART, and a large variety of non-site-specific elements such as Jockey, F, Doc, BS, G and Helena, although for these last two no potentially active copy has been identified. Our analysis has revealed one additional member of the Jockey clade, the JuanDm element.
Jockey, F and Doc elements
Jockey, F and Doc are very similar in sequence and probably descend from a common ancestor [19,20]. Our analyses confirm previous studies. These families include both full-size and variously defective copies. The copy number of Jockey, F and Doc was estimated to be around 50-100 by early studies [21,22,23]. About half of these copies are located near the chromocenter. This estimate has been refined to 31.60 ± 7.51 sites on chromosomal arms for Jockey, 31.40 ± 10 for F, and 26.20 ± 4.74 for Doc [24]. Our analyses reveal that each of these families in fact contains a very large total number of copies: at least 86 for Jockey, 94 for F and 128 for Doc. These are underestimates, as a large fraction of the heterochromatic part of the genome is not represented in the databases, and these regions are well known to contain transposable elements. We have identified seven full-size copies of Jockey, all surrounded by target site duplications (TSD) and mapping on chromosomal arms. The F and Doc families contain as many as 14 and 22 full-size copies, respectively. Surprisingly many full-size copies of F do not appear to be flanked by TSD. Large subsets of the defective copies of these families are truncated at the 5' end.
The Jockey family was reported to contain a subset of 3 kb long internally deleted elements [19,25]. Our analyses provide evidence for only two internally deleted elements of 2.6 and 2.9 kb in the genome of the y; cn bw sp strain. A subfamily of internally deleted elements might be specific for some strains, or might be confined to heterochromatic regions that are not represented in the databases.
G elements
No potentially active G element was reported by earlier studies. Only one complete G element was previously described, and it did not code for full-length ORF1 and ORF2 products [26]. However, some characteristic domains were recognized, such as cysteine-rich motifs of ORF1 and the reverse transcriptase domain of ORF2. The chromosomal distribution of G elements appeared to be fairly stable between strains. They were found mostly in tandem arrays in the nontranscribed spacer sequence of rDNA units [26]. Our analyses reveal 37 copies of G elements, most of which were short and variously deleted. We identified only one full-size G element surrounded by TSD, but it could not encode the complete ORF1 and ORF2 proteins. However, as this full-size element is not present in the sequences released by BDGP/EDGP, we cannot decide whether the fact that it appears to be unable to code for these products results from sequencing errors or if it is an inactive element. It is located in region 60E12-60F2 of the right arm of the second chromosome and is surrounded by other defective G elements organized in tandem repeats. In fact, the sequences of this G element and surrounding DNA are very similar to those previously described [26], indicating that it is the same inactive element.
TART elements
TART elements insert preferentially at the tips of the chromosomes with HeT-A elements and constitute the telomeres of the Drosophila chromosomes [27,28]. As the telomeric regions are not represented in the released sequences we did not expect to pull out TART or HeT-A elements. Not surprisingly, we have identified only 11 sequences matching TART reverse transcriptase, and none of them encodes a large protein. We found only some short regions with less than 60% identity to HeT-A. This emphasizes the idea that TART and HeT-A elements have a strong preference for telomeric regions.
BS elements
The number of copies of BS elements was estimated to be around five to ten [25,29]. Although the first two copies were identified as recent insertions within a gypsy element, they seem to transpose very infrequently because their distribution pattern is highly conserved between strains as judged by Southern blot analyses [25]. Our analyses reveal 19 copies of BS in the genome, only two of which are full size and surrounded by TSD.
Helena elements
Helena elements are non-LTR retrotransposons related to BS elements. They exist in various Drosophila species [30,31,32]. Hybridization studies have revealed sequences homologous to the D. virilis Helena element close to the chromocenter of D. melanogaster, and one copy was partly sequenced (GenBank accession code AF012030). We have found nine sequences matching Helena reverse transcriptase. No full-size copy is recognizable. They are unable to encode a large protein and the mean genetic distance between the different copies is very high. Presumably, these are the remnants of very ancient elements related to Helena, present in D. melanogaster or an ancestor species, and now extinct in D. melanogaster.
Some of the sequences found in our HMM search are equally distant from Helena and BS (52% identity). They possibly represent a new family, denoted Helena/BS in Figure 1. We noticed after this work was completed that a new non-LTR retrotransposon has recently been identified. The X element (GenBank accession code AF237761) belongs to the Helena/BS family.
JuanDm elements
We have identified one new family of non-LTR retrotransposons belonging to the Jockey clade, which we have called JuanDm because of its close relationship with Juan-A in Aedes aegypti [33] and Juan-C in Culex pipiens [34]. There are only eight JuanDm elements in the sequenced genome of the strain y; cn bw sp. One is only partially sequenced. The other seven are all surrounded by TSD (Figure 3). Four are full size, two are 5' truncated, and one has a 1.2 kb internal deletion. One of the full-size and one of the 5'-truncated JuanDm elements are also found in the sequences of BDGP/EDGP. There are five nucleotide differences between the sequences of the full-size copy of JuanDm from Release 1 and from BDGP/EDGP, including one base insertion and one base deletion, presumably resulting from sequencing errors. The complete JuanDm from BDGP/EDGP contains two overlapping ORFs with the capacity for encoding proteins very similar to those potentially encoded by Juan-A and Juan-C ORF1 and ORF2 (Figure 4).
Figure 3.
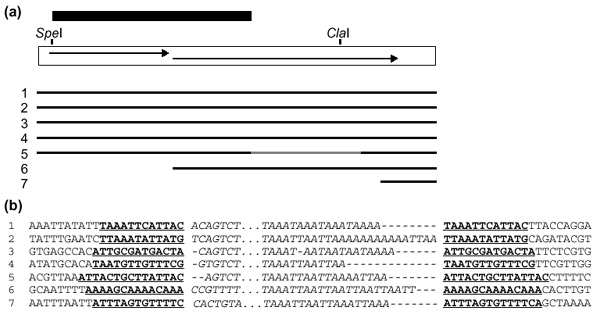
Structure of JuanDm elements in strain y; cn bw sp. (a) The full-size JuanDm element is represented as a white box, with two overlapping ORFs indicated as arrows. Positions of SpeI and ClaI restriction sites are shown and the black box above the element indicates the PCR fragment used as a probe in Figure 5a. Thick lines below represent the seven completely sequenced JuanDm elements found in Release 1. The internal deletion in element 5 is indicated as a gray region. (b) Sequences of integration sites of JuanDm elements shown in (a). Target site duplications are shown in bold and underlined. The 5'-most and 3'-most nucleotides of each element are shown in italics and separated by dots. For full-length elements, alignments in 5' region have been made to show variation in the 5' junction. The sequences of elements correspond to the following coordinates in release 1: 1, 13235891-13240123 in AE002787; 2, reverse complement of 7828981-7833212 in AE002566; 3, 4591855 4596087 in AE002593; 4, 268307-272538 in AE002629; 5, reverse complement of 7363833-7366823 in AE002708; 6, 874492-877244 in AE002638; 7, 2582603-2583169 in AE002602.
Figure 4.

Alignments of complete amino acid sequences of Juan ORF1 and ORF2. (a) ORF1; (b) ORF2. Identical residues are in highlighted in black, conserved residues are shaded in dark gray, and similar residues are shaded in light gray. Alignments and shadings were performed with VectorNTI software. The sequence of Juan-A in Aedes aegypti is from accession number M95171, the sequence of Juan-C in Culex pipiens is from M91082, the sequence of JuanDm in D. melanogaster is from AC005452 (coordinates 3425-7660).
There is very low heterogeneity between the eight JuanDm elements, which are more than 98% identical to each other at the DNA level. The JuanDm family is therefore a very young family in the genome of D. melanogaster. It is also present in other species from the D. melanogaster subgroup [35]: Figure 5a shows the result of hybridization of a JuanDm-specific DNA probe (Figure 3) to a Southern blot of genomic DNA digested with SpeI and ClaI. The probe reveals a JuanDm internal SpeI-ClaI fragment of 3 kb. A 3 kb fragment is observed in all species studied from the D. melanogaster subgroup: D. melanogaster, D. simulans, D. mauritiana, D. teissieri and D. yakuba. The strong signal intensity indicates that this fragment is present in multiple copies in D. melanogaster, D. simulans, D. mauritiana and D. yakuba. In addition, a few (two or three) fragments of various sizes are detected in these species. They might correspond to deleted elements. This indicates that the JuanDm family in these species is composed of very homogeneous elements. By contrast, in D. teissieri a 3 kb fragment is detected with a much lower intensity than in the other species. At least seven fragments of different sizes are also observed in this species. This suggests that the 3 kb internal fragment of JuanDm is not conserved in D. teissieri, and/or that these elements are more heterogeneous in size in D. teissieri than in other species of the D. melanogaster subgroup. No signal is detected in the more distant species D. virilis [35].
Figure 5.
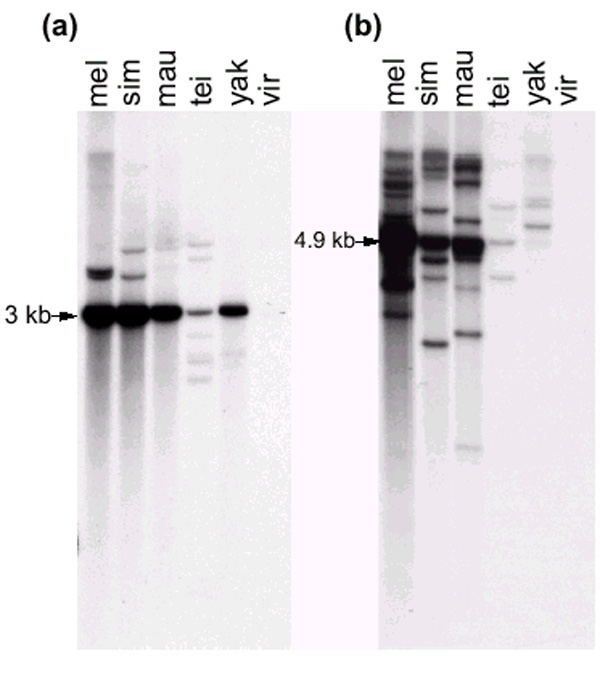
Southern blot analyses of JuanDm and You in various Drosophila species. Each lane contains 3 μg genomic DNA of the Drosophila melanogaster Cha strain (mel), D. simulans (sim), D. mauritiana (mau), D. teissieri (tei), D. yakuba (yak) and D. virilis (vir). (a) The genomic DNA was digested with SpeI and ClaI and hybridized with a JuanDm-specific probe (see Figure 3). (b) The genomic DNA was digested with SmaI and SacI and hybridized with a You-specific probe (see Figure 6).
Elements of the I clade
Until now the I element was the only member of the I clade to have been identified in D. melanogaster. This element has been extensively studied because it is responsible for the IR system of hybrid dysgenesis [36]. We have identified You, a new element of the I clade.
I elements
Studies of the I element family have been recently reviewed [37,38]. It is known that active I elements, also called I factors, are present only in some strains that are called inducer strains. Inducer strains are expected to contain five to ten full-size I elements in euchromatic regions, and about 30 defective elements mostly localized within pericentromeric heterochromatin [24,36,39,40]. The isogenic y; cn bw sp strain is inducer. Six full-size I elements can be identified in the sequences of Release 1. There is probably a seventh one for which sequences are available for both the 5' and 3' ends but not the middle. None of these elements has the coding capacities of an active I factor [41,42]. We assume that this is due to sequencing errors. All these copies are surrounded by TSD. They map on euchromatic sites. We have also identified six 5' truncated elements that are surrounded by TSD, and eight that are not. In addition, about 30 elements more divergent from active I elements are found. None of those is full size. Some are 5' truncated, some 3' truncated, some are truncated at both ends, and many have internal deletions. They are usually within unallocated scaffolds, strongly suggesting that they are in heterochromatic regions. Those that possess the 3' end of I terminate with a sequence related to TAA (TAAA)n instead of the regular TAA repeats of the active I elements. This termination was shown to be characteristic of I elements localized in pericentromeric heterochromatin [39].
You elements
The You family was first identified in the sequences of BDGP/EDGP on the basis of similarity between the DNA sequence of a full-size copy and that of the I element (58% all along the elements). Three full-size copies and one 5'-truncated copy surrounded by TSD can be found in the sequences of Release 1 (Figure 6). None of the complete You elements can produce full products of ORF1 and ORF2. This is presumably because of sequencing errors as one of these copies was also sequenced by BDGP/EDGP, revealing full coding capacities. You and I are closely related: they both encode very similar ORF1 products containing three cysteine-rich motifs (Figure 7). The products of ORF2 are also very similar and contain endonuclease, reverse transcriptase and RNase H domains as well as one cysteine-rich motif. You elements terminate at the 3' end with A/T rich sequences.
Figure 6.

Structure of You elements in strain y; cn bw sp. (a) The full-size You element is represented as a white box, with the two ORFs indicated as arrows. Positions of SmaI and SacI restriction sites are shown and the black box above the element indicates the PCR fragment used as a probe in Figure 5b. Thick lines below represent full-size (1-3) and 5' truncated (4) You elements found in Release 1. The internal deletion in element 3 is indicated as a gray region. (b) Sequences of integration sites of You elements shown in (a). Target site duplications are shown in bold and underlined. The most 5' and 3' nucleotides of each element are shown in italics and separated by dots. For full-length elements, alignments in 5' region have been made to show variation in the 5' junction. The sequences of elements corespond to following coordinates in release 1: 1, 56440-61816 in AE002620; 2, 2419411-2424785 in AE002647; 3, reverse complement of 1119136-1124111 in AE002566; 4, reverse complement of 5105-6524 in AE002601.
Figure 7.

Alignments of complete amino acid sequences from You and I elements ORF1 and ORF2. (a) ORF1; (b) ORF2. Identical residues are highlighted in black, similar residues are in light gray. Alignment and shading was performed with VectorNTI software. The sequence of I is from accession number M14954 corrected according to [42], the sequence of You is from AL03893 (coordinates 1-5255) and AL022018 (coordinates 38180-38397) assembled manually.
Strikingly, we have noticed that the six You elements that could be localized, map in distal or proximal regions of chromosomal arms: 1F and 19E-F on chromosome X, 39E and 40A on chromosome II, and 60F and 79E-F on chromosome III. This is a very unusual distribution, and it would be interesting to determine whether You elements are also localized similarly in other D. melanogaster strains. Examining the sequences surrounding You elements did not reveal an obvious bias for insertion sites. Their unusual distribution is therefore unlikely to be the result of sequence preference for insertion. Alternatively, euchromatic copies of You could be the few survivors of a former broader You family that is in the process of being eliminated from the genome by stochastic loss, as in the case of Drosophila mariner elements [43]. Their confinement near the frontiers between euchromatin and heterochromatin could reflect a lower rate of loss of sequences in these regions.
The You family is also present in other species of the D. melanogaster subgroup [35]: Figure 5b shows the result of hybridization of a You-specific DNA probe (Figure 6) to a Southern blot of genomic DNA digested with SmaI and SacI. The probe reveals an internal SmaI-SacI fragment of 4.9 kb. A 4.9 kb band hybridizing strongly to the probe is observed in D. melanogaster and in sibling species D. simulans and D. mauritiana, indicating that these species contain several copies of potentially full-size You elements. The more distant species D. teissieri and D. yakuba show much weaker signals, and the presence of the 4.9 kb fragment is not certain. These species contain sequences related to You, but these sequences are divergent from those of the D. melanogaster You elements. Finally, no hybridization signal is detected in D. virilis, which is outside the D. melanogaster subgroup [35], even with longer exposures.
Elements of the R1 clade
Most non-LTR retrotransposons of the R1 clade are site-specific: R1Dm inserts preferentially at one site into the rDNA units. Recently, we have described two new subfamilies of elements - Waldo-A and Waldo-B - that belong to the R1 clade and do not seem to have strong insertion site preference.
Waldo-A and Waldo-B elements
The Waldo-A and Waldo-B families were first identified by analyzing the sequences released by BDGP/EDGP and are described elsewhere [44]. Members of both subfamilies are found in the present study. There are two full-size Waldo-A and five full-size Waldo-B elements. Most of them are surrounded by TSD. All 5'-truncated Waldo-A elements are surrounded by TSD and are very similar to complete Waldo-A elements (>98% identity at the DNA level). Only a minority of Waldo-B 5'-truncated elements is surrounded by TSD. In addition variously defective elements with less than 98% identity to Waldo-A or Waldo-B elements have been found.
R1Dm elements
Only fragments of elements with similarity to R1Dm have been found in our search, presumably because R1Dm inserts preferentially within rDNA repeats, which are not represented in the databases.
Elements of other clades
Partial R2Dm elements were identified in the BDGP/EDGP sequences but not in Release 1, presumably for the same reasons as for R1Dm.
We have found sequences that match the reverse transcriptase of the non-LTR retrotransposons Q in Anopheles gambiae [45] and LOA in D. silvestris [46]. These non-LTR retrotransposons belong to the LOA and the CR1 clades, respectively, which were supposed to have no representatives in D. melanogaster. We have also found some short sequences showing weak similarities to the element Bilbo in D. subobscura [47], which also belongs to the LOA clade. As in the case of Helena, these sequences are presumably remnants of very ancient elements from the LOA and CR1 clades, once present in D. melanogaster or an ancestor and now extinct.
Discussion
Put together, the sequences of BS, Doc, F, G, I, Jockey, JuanDm, Waldo-A, Waldo-B and You elements presented in this work make 1134 kb, which is almost 1% of the 120 Mb of Release 1 [14]. This percentage would certainly increase if our analysis could be extended to all heterochromatic portions of the genome. In particular pericentromeric heterochromatin regions are known to be enriched in defective transposable elements that are thought to be old components of the genome [12,13]. Only a small subset of the sequences in Release 1 (3.8 Mb) presumably originate from these regions because they could not be localized on chromosomal arms [14]. Many of the defective copies of non-LTR retrotransposons were found in these unlocalized sequences.
The mean genetic distance between members of a given family reflects their degree of heterogeneity. Assuming that non-LTR retrotransposons of different families accumulate mutations at the same rate, elements belonging to old families are expected to be more heterogeneous than elements of recent families. This reasoning does not take into account, however, possible bursts of invasion of a genome by a particular element or subset of elements of one family. This situation is well documented in the case of the I element family [37]. Indeed, active I elements that are now present in D. melanogaster have invaded the species very recently, during the 20th century. However, all D. melanogaster strains contain defective heterochromatic I elements that display an average of 94% sequence identity with each other and with the I factor [40]. These are very old remnants of former active I elements that were present in the common ancestor of all species from the D. melanogaster subgroup and were lost in the ancestor of the D. melanogaster species lineage. It is therefore not inconceivable that this kind of event may also have occurred in the case of other non-LTR retrotransposon families. When possible, we have estimated the genetic distance between only the subset of longest available elements within a family. This was done for the Doc, F, G, I, Jockey, JuanDm, Waldo-A, Waldo-B and You families (data not shown). Not surprisingly, the subset of longest elements is constantly more homogeneous than the whole family, indicating that the longest elements are probably still active or result from recent inactivation. One should keep in mind, however, that the error rate in the repeated sequences in Release 1 is not negligible and therefore the degree of heterogeneity observed in a small number of copies is probably overestimated.
The high rate of errors in repetitive sequences of Release 1 renders thorough analysis of transposable element families uncertain. For example, on the basis of the recognition of reverse transcriptase coding capacities, You elements were not recognized in the sequences of Release 1 (Figure 1a) but were identified in the BDGP/EDGP sequences (Figure 1b). It is possible that some non-LTR retrotransposon families have been missed by our study. More accurate sequences of repetitive DNA regions are expected in the near future [17]. They will provide invaluable material for the thorough phylogenetic analyses of families of transposable elements. In particular, analysis of the sequences and organization of the different copies of a given family will provide information useful in understanding the processes by which these elements evolve in the genome.
The annotations of the sequences of Release 1 are in progress. Most copies of non-LTR retrotransposons that we have found in the present study are not annotated yet. The coordinates of all sequences identified in our studies are available as an additional data file with the online version of this paper.
Materials and methods
Identification of reverse transcriptase sequences
Drosophila melanogaster sequences produced by Celera [14] and the BDGP/EDGP [16] were used in the analysis. A six-frame translation of all the sequences was produced. To reduce the size of the data set to be analyzed by time-consuming HMM software, only amino-acid sequences containing a motif [FY]XDD, which is conserved among reverse transcriptases [15], were extracted for the analysis. The HMMER 2.1.1 software [48] was used to identify all sequences in this subset matching the full-length model of reverse transcriptase, which was built using a seed alignment of reverse transcriptase sequences (accession number PF00078) obtained from the Pfam database [49]. Only matches with scores above zero were considered in the analysis.
Handling of sequences was facilitated by scripts from the SEALS package [50]. The results of HMMER searches were analyzed using scripts that we designed specially, grouped into families and classified on the basis of their similarities to known retrotransposons. The relationships between the families of non-LTR retrotransposons were determined by making neighbor-joining trees using the CLUSTAL W software [51].
Analysis of non-LTR retrotransposon families
BLASTN searches were performed in sequences of Release 1 and of BDGP/EDGP using WU-BLAST 2.0 package [52] and full-length elements from different families as queries. The results were analyzed using scripts that were especially written and based on the BioPerl package [53]. All high-scoring pairs (HSPs) with percentage identities greater than 70% and lengths greater than 200 nucleotides were used to define distinct copies in the same family. These limiting values were arbitrarily chosen to take into account all diverged and truncated copies in a family while filtering out doubtful hits. HSPs in the same hit were checked manually for overlaps or internal deletions relative to a full-length element and joined together when necessary. Genomic coordinates for all copies were determined in this way and element sequences with flanking regions were extracted for further analysis. Target site duplications were searched in a semi-manual manner with the aid of bl2seq program from the NCBI BLAST package [54]. Divergence of elements within a family was determined in the following manner: the longest element sequence in a family was used in BLAST searches against all other sequences in the family and resulting BLAST output was converted into multiple alignment with the MView program [55]. The alignment obtained was used to calculate genetic distances between copies in a family by CLUSTAL W software. Divergence of a family was determined as a mean genetic distance between the longest element and all the other elements in a family. The error rate was estimated by comparing sequences of full-length Jockey and JuanDm elements which have the same flanking sequences (100-200 bp flanks were used) in both Release 1 and BDGP/EDGP databases.
Southern blots
Digestion of genomic DNA, gel electrophoresis, transfer on NytranN nylon membranes (Schleicher and Schuell) and hybridization with 32P-labeled DNA probes were performed following standard procedures [56] and suppliers' specifications. Hybridizations were carried out overnight at 42°C in 50% formamide. Washes were in 2x standard sodium sulfate (SSC), 0.1% sodium dodecyl sulfate (SDS) followed by 0.1 × SSC, then 0.1% SDS at 42°C. The DNA fragments used as probes were obtained by PCR amplifications from the isogenic y; cn bw sp strain genomic DNA using standard conditions with Taq DNA polymerase (Promega). For the JuanDm probe oligonucleotides 5'-AAGGAATAAACCACTAGTGGAGCGCC-3' and 5'-CAGGGAGTGTAAGCTTGGAGTGATG-3' were used as primers. For the You probe oligonucleotides 5'-GATCTTCTTATCAACGCGTACGTGC-3' and 5'-CCCAGGAGTATTGTGGATCCGTTAAG-3' were used as primers.
Additional data
The following additional data are included with the online version of this paper: the coordinates of all sequences identified in this study.
Supplementary Material
The coordinates of all sequences identified in this study
Acknowledgments
Acknowledgements
We thank all who contributed open-source software used in this project (BioPerl, HMMER, SEALS, Clustal W and MView), Alexander Blinov for providing resources and support in the computer research, Ael Laglaine for help in the study of You elements, Christophe Terzian for helpful advice and Stephen Wicks for assistance with manuscript preparation. This work was supported by grants from the Russian Foundation for Basic Research (RFBR), from the Centre National de la Recherche Scientifique (CNRS) and from the Association pour la Recherche sur le Cancer (ARC).
References
- Eickbush TH. Transposing without ends: the non-LTR retrotransposable elements. New Biol. 1992;4:430–440. [PubMed] [Google Scholar]
- Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Eickbush TH. The site-specific ribosomal DNA insertion element R1Bm belongs to a class of non-long-terminal-repeat retrotransposons. Mol Cell Biol. 1988;8:114–123. doi: 10.1128/mcb.8.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubczak JL, Xiong Y, Eickbush TH. Type I (R1) and type II (R2) ribosomal DNA insertions of Drosophila melanogaster are retrotransposable elements closely related to those of Bombyx mori. J Mol Biol. 1990;212:37–52. doi: 10.1016/0022-2836(90)90303-4. [DOI] [PubMed] [Google Scholar]
- Xiong YE, Eickbush TH. Functional expression of a sequence-specific endonuclease encoded by the retrotransposon R2Bm. Cell. 1988;55:235–246. doi: 10.1016/0092-8674(88)90046-3. [DOI] [PubMed] [Google Scholar]
- Yang J, Malik HS, Eickbush TH. Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci USA. 1999;96:7847–7852. doi: 10.1073/pnas.96.14.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Maranon C, Olivares M, Alonso C, Lopez MC. Characterization of a non-long terminal repeat retrotransposon cDNA (L1Tc) from Trypanosoma cruzi: homology of the first ORF with the ape family of DNA repair enzyme. J Mol Biol. 1995;247:49–59. doi: 10.1006/jmbi.1994.0121. [DOI] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Malik HS, Eickbush TH. NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics. 2000;154:193–203. doi: 10.1093/genetics/154.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Smit AF. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;6:743–748. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- Vaury C, Bucheton A, Pelisson A. The beta heterochromatic sequences flanking the I elements are themselves defective transposable elements. Chromosoma. 1989;98:215–224. doi: 10.1007/BF00329686. [DOI] [PubMed] [Google Scholar]
- Dimitri P, Junakovic N. Revising the selfish DNA hypothesis: new evidence on accumulation of transposable elements in heterochromatin. Trends Genet. 1999;15:123–124. doi: 10.1016/s0168-9525(99)01711-4. [DOI] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990;9:3353–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkeley Drosophila Genome Project http://www.fruitfly.org
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Celera Analysis of the Drosophila Genome http://www.celera.com/genomeanalysis
- Priimagi AF, Mizrokhi LJ, Ilyin YV. The Drosophila mobile element jockey belongs to LINEs and contains coding sequences homologous to some retroviral proteins. Gene. 1988;70:253–262. doi: 10.1016/0378-1119(88)90197-7. [DOI] [PubMed] [Google Scholar]
- O'Hare K, Alley MR, Cullingford TE, Driver A, Sanderson MJ. DNA sequence of the Doc retroposon in the white-one mutant of Drosophila melanogaster and of secondary insertions in the phenotypically altered derivatives white-honey and white-eosin. Mol Gen Genet. 1991;225:17–24. doi: 10.1007/BF00282637. [DOI] [PubMed] [Google Scholar]
- Mizrokhi LJ, Obolenkova LA, Priimägi AF, Ilyin YV, Gerasimova TI, Georgiev GP. The nature of unstable mutations and reversions in the locus cut of Drosophila melanogaster: molecular mechanism of transposition memory. EMBO J. 1985;4:3781–3787. doi: 10.1002/j.1460-2075.1985.tb04148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawid IB, Long EO, DiNocera PP, Pardue ML. Ribosomal insertion-like elements in Drosophila melanogaster are interspersed with mobile sequences. Cell. 1981;25:399–408. doi: 10.1016/0092-8674(81)90058-1. [DOI] [PubMed] [Google Scholar]
- Vaury C, Chaboissier MC, Drake ME, Lajoinie O, Dastugue B, Pelisson A. The Doc transposable element in Drosophila melanogaster and Drosophila simulans: genomic distribution and transcription. Genetica. 1994;93:117–124. doi: 10.1007/BF01435244. [DOI] [PubMed] [Google Scholar]
- Vieira C, Lepetit D, Dumont S, Biemont C. Wake up of transposable elements following Drosophila simulans worldwide colonization. Mol Biol Evol. 1999;16:1251–1255. doi: 10.1093/oxfordjournals.molbev.a026215. [DOI] [PubMed] [Google Scholar]
- Campuzano S, Balcells L, Villares R, Carramolino L, Garcia-Alonso L, Modolell J. Excess function hairy-wing mutations caused by gypsy and copia insertions within structural genes of the achaete-scute locus of Drosophila. Cell. 1986;44:303–312. doi: 10.1016/0092-8674(86)90764-6. [DOI] [PubMed] [Google Scholar]
- Di Nocera PP, Graziani F, Lavorgna G. Genomic and structural organization of Drosophila melanogaster G elements. Nucleic Acids Res. 1986;14:675–691. doi: 10.1093/nar/14.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis RW, Ganesan R, Houtchens K, Tolar LA, Sheen FM. Transposons in place of telomeric repeats at a Drosophila telomere. Cell. 1993;75:1083–1093. doi: 10.1016/0092-8674(93)90318-k. [DOI] [PubMed] [Google Scholar]
- Sheen FM, Levis RW. Transposition of the LINE-like retrotransposon TART to Drosophila chromosome termini. Proc Natl Acad Sci USA. 1994;91:12510–12514. doi: 10.1073/pnas.91.26.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udomkit A, Forbes S, Dalgleish G, Finnegan DJ. BS a novel LINE-like element in Drosophila melanogaster. Nucleic Acids Res. 1995;23:1354–1358. doi: 10.1093/nar/23.8.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA, Schutzman JL, Hartl DL, Lozovskaya ER. Diverse transposable elements are mobilized in hybrid dysgenesis in Drosophila virilis. Proc Natl Acad Sci USA. 1995;92:8050–8054. doi: 10.1073/pnas.92.17.8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA, Lozovskaya ER, Hartl DL. High intrinsic rate of DNA loss in Drosophila. Nature. 1996;384:346–349. doi: 10.1038/384346a0. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Hartl DL. High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol. 1998;15:293–302. doi: 10.1093/oxfordjournals.molbev.a025926. [DOI] [PubMed] [Google Scholar]
- Mouches C, Bensaadi N, Salvado JC. Characterization of a LINE retroposon dispersed in the genome of three non-sibling Aedes mosquito species. Gene. 1992;120:183–190. doi: 10.1016/0378-1119(92)90092-4. [DOI] [PubMed] [Google Scholar]
- Agarwal M, Bensaadi N, Salvado JC, Campbell K, Mouches C. Characterization and genetic organization of full-length copies of a LINE retroposon family dispersed in the genome of Culex pipiens mosquitoes. Insect Biochem Mol Biol. 1993;23:621–629. doi: 10.1016/0965-1748(93)90036-r. [DOI] [PubMed] [Google Scholar]
- Lachaise D, Cariou M-L, David JR, Lemenier F, Tsacas L, Ashburner M. Historical biogeography of the Drosophila melanogaster species subgroup. In Evolutionary Biology Edited by Hecht MK, Wallace B, Prance GT Plenum Publishing Corporation; 1988;22:152–225. [Google Scholar]
- Bucheton A, Paro R, Sang HM, Pelisson A, Finnegan DJ. The molecular basis of I-R hybrid dysgenesis in Drosophila melanogaster: identification, cloning, and properties of the I factor. Cell. 1984;38:153–163. doi: 10.1016/0092-8674(84)90536-1. [DOI] [PubMed] [Google Scholar]
- Bucheton A, Vaury C, Chaboissier MC, Abad P, Pelisson A, Simonelig M. I elements and the Drosophila genome. Genetica. 1992;86:175–190. doi: 10.1007/BF00133719. [DOI] [PubMed] [Google Scholar]
- Bucheton A, Busseau I, Teninges D. I elements in Drosophila melanogaster. In Mobile DNA II Edited by Craig N, Craigie R, Gellert M, Lambowitz A Washington, DC: American Society for Microbiology; 2000.
- Crozatier M, Vaury C, Busseau I, Pelisson A, Bucheton A. Structure and genomic organization of I elements involved in I-R hybrid dysgenesis in Drosophila melanogaster. Nucleic Acids Res. 1988;16:9199–9213. doi: 10.1093/nar/16.19.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaury C, Abad P, Pelisson A, Lenoir A, Bucheton A. Molecular characteristics of the heterochromatic I elements from a reactive strain of Drosophila melanogaster. J Mol Evol. 1990;31:424–431. doi: 10.1007/BF02106056. [DOI] [PubMed] [Google Scholar]
- Fawcett DH, Lister CK, Kellett E, Finnegan DJ. Transposable elements controlling I-R hybrid dysgenesis in D. melanogaster are similar to mammalian LINEs. Cell. 1986;47:1007–1015. doi: 10.1016/0092-8674(86)90815-9. [DOI] [PubMed] [Google Scholar]
- Abad P, Vaury C, Pelisson A, Chaboissier MC, Busseau I, Bucheton A. A long interspersed repetitive element - the I factor of Drosophila teissieri is able to transpose in different Drosophila species. Proc Natl Acad Sci USA. 1989;86:8887–8891. doi: 10.1073/pnas.86.22.8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Moriyama EN, Lidholm DA, Hartl DL. Horizontal transmission, vertical inactivation, and stochastic loss of mariner-like transposable elements. Mol Biol Evol. 1995;12:62–72. doi: 10.1093/oxfordjournals.molbev.a040191. [DOI] [PubMed] [Google Scholar]
- Busseau I, Berezikov E, Bucheton A. Identification of Waldo-A and Waldo-B, two closely related non-LTR retrotrans-posons in Drosophila. Mol Biol Evol. 2001. [DOI] [PubMed]
- Besansky NJ, Bedell JA, Mukabayire O. Q: a new retrotransposon from the mosquito Anopheles gambiae. Insect Mol Biol. 1994;3:49–56. doi: 10.1111/j.1365-2583.1994.tb00150.x. [DOI] [PubMed] [Google Scholar]
- Felger I, Hunt JA. A non-LTR retrotransposon from the Hawaiian Drosophila: the LOA element. Genetica. 1992;85:119–130. doi: 10.1007/BF00120318. [DOI] [PubMed] [Google Scholar]
- Blesa D, Martinez-Sebastian MJ. bilbo, a non-LTR retrotransposon of Drosophila subobscura: a clue to the evolution of LINE-like elements in Drosophila. Mol Biol Evol. 1997;14:1145–1153. doi: 10.1093/oxfordjournals.molbev.a025724. [DOI] [PubMed] [Google Scholar]
- Profile Hidden Markov Models for Biological Sequence Analysis http://hmmer.wustl.edu/
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEALS Home Page http://www.ncbi.nlm.nih.gov/Walker/SEALS
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington University BLAST Archives http://blast.wustl.edu
- The Bioperl Project http://bio.perl.org
- NCBI BLAST Ftp site ftp://ncbi.nlm.nih.gov/blast/
- Brown NP, Leroy C, Sander C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics. 1998;14:380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, a Laboratory Manual Cold Spring Harbor, New York: 2nd edition Cold Spring Harbor Laboratory Press; 1989.
- Kumar S, Tamura K, Nei M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput Appl Biosci. 1994;10:189–191. doi: 10.1093/bioinformatics/10.2.189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The coordinates of all sequences identified in this study