

Abstract
Our abilities to detect and enumerate pollutant-biodegrading microorganisms in the environment are rapidly advancing with the development of molecular genetic techniques. Techniques based on multiplex and real-time PCR amplification of aromatic oxygenase genes were developed to detect and quantify aromatic catabolic pathways, respectively. PCR primer sets were identified for the large subunits of aromatic oxygenases from alignments of known gene sequences and tested with genetically well-characterized strains. In all, primer sets which allowed amplification of naphthalene dioxygenase, biphenyl dioxygenase, toluene dioxygenase, xylene monooxygenase, phenol monooxygenase, and ring-hydroxylating toluene monooxygenase genes were identified. For each primer set, the length of the observed amplification product matched the length predicted from published sequences, and specificity was confirmed by hybridization. Primer sets were grouped according to the annealing temperature for multiplex PCR permitting simultaneous detection of various genotypes responsible for aromatic hydrocarbon biodegradation. Real-time PCR using SYBR green I was employed with the individual primer sets to determine the gene copy number. Optimum polymerization temperatures for real-time PCR were determined on the basis of the observed melting temperatures of the desired products. When a polymerization temperature of 4 to 5°C below the melting temperature was used, background fluorescence signals were greatly reduced, allowing detection limits of 2 × 102 copies per reaction mixture. Improved in situ microbial characterization will provide more accurate assessment of pollutant biodegradation, enhance studies of the ecology of contaminated sites, and facilitate assessment of the impact of remediation technologies on indigenous microbial populations.
The adoption of molecular genetic techniques has greatly improved our ability to characterize microbial populations in situ, leading to more accurate assessments of pollutant biodegradation and the impact of remediation technologies. Initially, biases associated with culturing were avoided by using DNA hybridization techniques with probes containing the catabolic gene of interest. Cross-hybridization at low stringency (46) and exclusion of related but not identical genes at high stringency (15, 30, 56) may produce false-positive and false-negative results with environmental samples. PCR amplification with primers based on conserved regions within the gene of interest, however, will permit amplification despite variability in the overall sequence and detect genotypes not detected by hybridization alone. Molecular methods, particularly in quantitative PCR (Q-PCR), continue to advance rapidly. Our laboratory described a competitive Q-PCR technique to enumerate catechol 2,3-dioxygenase genes as a means to evaluate bioremediation of aromatic hydrocarbons at petroleum-contaminated sites (35). Here we present primer sets targeting additional aromatic catabolic genes and the use of real-time PCR for quantification to extend our ability to enumerate genotypes involved in biodegradation of aromatic pollutants.
The two main approaches to Q-PCR are competitive and real-time PCR. In competitive PCR, a comparison of the intensities of the standard and target amplicons allows quantification (42). One limitation of this method is that amplification efficiency of the target and standard are not always equivalent. Furthermore, post-PCR analysis of the products is labor-intensive and not amenable to the large-scale screening of environmental samples. Real-time PCR offers an accurate, sensitive method of quantification without the labor-intensive postamplification analysis and assumptions required by competitive PCR. Detection of product during amplification can be accomplished by several means, including molecular beacons (55), hybridization probes (60), and BODIPY FL-labeled probes and primers (27), but hydrolysis probes and DNA-binding dyes are the most commonly reported methods. The hydrolysis probe (TaqMan) assay takes advantage of the 5′-nuclease ability of DNA polymerase to hydrolyze a labeled probe bound to its target amplicon to produce a signal. The SYBR green assay relies on fluorescence signal produced as the dye binds to double-stranded DNA during the extension step. While the hydrolysis probe provides an additional degree of specificity, identifying an additional highly conserved region for the probe may not always be possible. Furthermore, both the probe and the primers must be from a highly conserved region of the target to avoid biasing quantitation (2). Simpler methods using DNA-staining dyes, such as SYBR green I, have been successful (36, 62), do not require the design of an internal hybridization probe, and thus can be used with any established PCR primers with only minor modifications of the described protocols.
This study was conducted to develop multiplex and real-time PCR procedures to quantify aromatic catabolic genes in environmental samples and eventually improve our understanding of biodegradation in the field. The large subunit of aromatic oxygenase genes was chosen as the indicator gene, because it has been implicated in substrate specificity and is one of the rate-limiting steps in aromatic hydrocarbon biodegradation (14) and its DNA sequence is conserved for oxygenases targeting the same substrate. Alignments were constructed from groups of related oxygenase genes, and each primer set was chosen from a conserved region unique to that group of oxygenases. Thus, a single primer set will detect an entire subfamily of related oxygenase genes rather than a species-specific catabolic gene. In all, PCR primer sets which targeted biphenyl dioxygenase, naphthalene dioxygenase, toluene dioxygenase, toluene/xylene monooxygenase, phenol monooxygenase, and ring-hydroxylating toluene monooxygenase genes were identified. Testing and optimization with genetically well-characterized bacterial strains demonstrated the specificity of each primer set. Multiplex PCR protocols were developed to permit simultaneous detection of aromatic oxygenase genes and facilitate rapid screening of environmental samples. Real-time PCR with SYBR green I was used to determine the gene copy number with a quantification limit of 2 × 102 copies of target per reaction mixture. The primer sets and real-time PCR methods presented will be used to assess natural attenuation, investigate the ecology of contaminated sites, and aid in optimization of bioremediation in the field.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used are listed in Table 1. Liquid cultures were grown overnight in minimal medium (34) supplemented with the appropriate carbon source with shaking (125 rpm) at 30°C. Biphenyl and naphthalene were added as solids to the liquid medium or to the lids of inverted agar plates. Toluene was provided as a gas in sealed containers (43). Burkholderia sp. strain DNT was grown as described by Suen et al. (49). Rhodococcus sp. strain RHA1, Rhodococcus erythropolis TA421, Pseudomonas aeruginosa JI104, and Pseudomonas mendocina KR1 were grown on C medium (31).
TABLE 1.
Aromatic oxygenase genes used to deduce conserved regions for PCR primers
| Gene | GenBank accession no. | Source organism | Reference or source |
|---|---|---|---|
| Biphenyl dioxygenase | |||
| bphA | U47637 | Comamonas testosteroni B-356 | 50 |
| bphA1 | D17319 | Pseudomonas sp. strain KKS102 | 25 |
| bphA1 | X80041 | Rhodococcus globerulus P6 | 1a |
| bpdC1 | U27591 | Rhodococcus sp. strain M5 | 57 |
| bphA1 | D88021 | Rhodococcus erythropolis TA421 | 26 |
| bphA | M86348 | Burkholderia sp. strain LB400 | 11 |
| bphA1 | M83673 | Pseudomonas pseudoalcaligenes KF707 | 51 |
| bphA1 | D32142 | Rhodococcus sp. strain RHA1 | 33 |
| bphA1 | AJ010057 | Burkholderia sp. strain JB1 | M. Dignum |
| bphA1 | U95054 | Pseudomonas sp. strain B4 | V. Durcrocq |
| Isopropylbenzene and ethylbenzene dioxygenases | |||
| ipbA1 | U24277 | Rhodococcus erythropolis BD2 | 24 |
| ipbA1 | U53507 | Pseudomonas sp. strain JR1 | 41 |
| ipbAa | AF006691 | Pseudomonas putida RE204 | 10 |
| cumA1 | D37828 | Pseudomonas fluorescens IP01 | 1 |
| edoA1 | AF049851 | Pseudomonas fluorescens CA-4 | 7 |
| Naphthalene dioxygenase | |||
| nahAc | M83949 | Pseudomonas putida G7 | 47 |
| doxB | M60405 | Pseudomonas sp. strain C18 | 8 |
| nahAc | U49496 | Pseudomonas sp. strain 9816-4 | J. Parales |
| ndoC2 | AF004284 | Pseudomonas putida ATCC 17484 | C. Hamann |
| pahAc | AB004059 | Pseudomonas putida OUS82 | 52 |
| pahA3 | D84146 | Pseudomonas aeruginosa PaK1 | N. Takizawa |
| nahA3 | AF010471 | Pseudomonas putida BS202 | 4 |
| nagAc | AF036940 | Pseudomonas sp. strain U2 | 13 |
| dntAc | U62430 | Burkholderia sp. strain DNT | 49 |
| phnAc | AF061751 | Burkholderia sp. strain RP007 | 29 |
| phnAc | AB024945 | Alcaligenes faecalis AFK2 | H. Kiyohara |
| nahAc | AF053737 | Cycloclasticus sp. strain 1P-32 | B. P. Hedlund |
| nahAc | AF053736 | Neptunomonas naphthovorans NAG-2N-126 | 19 |
| nahAc | AF053735 | Neptunomonas naphthovorans NAG-2N-113 | 19 |
| nahAc | AF093000 | Cycloclasticus sp. strain W | 16 |
| nahAc | AF092998 | Cycloclasticus pugetii PS-1 | 16 |
| narAc | AF082663 | Rhodococcus sp. strain NCIMB12038 | 28 |
| nahAc | AF039533 | Pseudomonas stutzeri AN10 | 5 |
| Toluene/benzene/chlorobenzene dioxygenase | |||
| todC1 | J04996 | Pseudomonas putida F1 | 63 |
| bedC1 | L04642 | Pseudomonas putida ML2 | 53 |
| tcbAa | U15298 | Pseudomonas sp. strain P51 | 58 |
| Toluene monooxygenase | |||
| xylM | D63341 | Pseudomonas putida mt-2 | U. Harayamal |
| xylM | AF019635 | Pseudomonas putida HS1 | D. Kunz |
| ntnM | AF043544 | Pseudomonas sp. strain TW3 | 21 |
| Ring hydroxylating monooxygenases | |||
| tmoF | M95045 | Pseudomonas mendocina KR1 | 61 |
| tbuA1 | U04052 | Ralstonia picketti PKO1 | 6 |
| tbhA | AF001356 | Burkholderia cepacia AA1 | 30a |
| tbmD | L40033 | Pseudomonas sp. strain JS150 | 22 |
| bmoA | Pseudomonas aeruginosa JI104 | ||
| Phenol hydroxylase | |||
| dmpN | M60276 | Pseudomonas putida CF600 | 39 |
| phhN | X79063 | Pseudomonas putida P35X | 38 |
| Phenol hydroxylase alpha subunit | AB016863 | Comamonas testosteroni R2 | H. Futamata |
| Phenol hydroxylase alpha subunit | AB016862 | Comamonas sp. strain E6 | H. Futamata |
| Phenol hydroxylase alpha subunit | AB016861 | Burkholderia cepacia E1 | H. Futamata |
| Phenol hydroxylase alpha subunit | AB016859 | Pseudomonas putida P-6 | H. Futamata |
| Phenol hydroxylase alpha subunit | AB016858 | Pseudomonas putida P-8 | H. Futamata |
| poxD | AF026065 | Ralstonia sp. strain E2 | 20 |
DNA extractions.
DNA extractions from pure cultures except Rhodococcus sp. strain RHA1 and R. erythropolis TA421 were performed using the FastDNA kit (Bio 101, Vista, Calif.) and the FP120 FastPrep cell disruptor (Savant Instruments, Inc., Holbrook, N.Y.). Strains TA421 and RHA1 were incubated with 2 mg of lysozyme per liter at 37°C for 1 h, and then total genomic DNA was isolated as described previously (32). DNA concentrations were quantified by fluorometry using a DNA fluorometer (model TKO100; Hoefer Scientific Instruments, San Francisco, Calif.) calibrated with calf thymus DNA.
Phylograms and alignments.
DNA and amino acid sequences of the large subunits of aromatic oxygenases were retrieved from GenBank (3) and aligned using Clustal W version 1.7 (54). Genes used for these alignments are given in Table 1. Phylograms were constructed with DNAMAN software programs (Lynnon BioSoft, Vaudreuil, Quebec, Canada) using the neighbor-joining method (45) and bootstrap analysis. Subfamilies of oxygenase genes were identified from the phylograms. Alignments of the sequences of these subfamilies were examined for conserved regions for use as PCR primers. To confirm that the chosen primer sets were unique, alignments were also constructed with related oxygenase genes. Chosen primer sets were also compared to sequences available on the GenBank database to further confirm that primer sequences were unique to the target. Primer Premier software (PREMIER Biosoft International, Palo Alto, Calif.) was used to screen potential primer sets for hairpins and ensure that primer pairs had similar annealing temperatures.
PCR primers and conditions.
PCR primers were chosen from conserved regions in the DNA sequences observed during alignment of each group of aromatic oxygenases. A description of the PCR primers (patent pending) and conditions is shown in Table 2. The following combinations of primers were also used for multiplex PCR: PHE/NAH, TOL/TOD, and BPH2/BPH4. Annealing temperatures for the three combinations of primers in multiplex PCR were 49, 55, and 62°C, respectively. All PCR mixtures contained 1× PCR buffer (Promega, Madison, Wis.), 0.2 mM each of the four deoxynucleoside triphosphates (Amersham Pharmacia, Piscataway, N.J.), and 1 U of Taq polymerase. Annealing temperature and DNA (10, 1, and 0.1 ng), MgCl2, and primer concentrations were optimized for each primer set. MgCl2 concentrations were increased from 1.5 to 3.0 mM until yield decreased or failed to increase. Primer concentrations were increased from 0.1 to 0.5 μM, except for BPH1 which was also tested at lower concentrations. Conventional and multiplex PCR was performed in a PTC-100 Programmable Thermal Controller (MJ Research, Inc., Waltham, Mass.) with the following temperature program: (i) 10 min at 95°C; (ii) 30 cycles, with 1 cycle consisting of 1 min at 95°C, 1 min at the optimum annealing temperature, and 2 min at 72°C, and (iii) a final extension step of 10 min at 72°C. All experiments included control reaction mixtures without added DNA. PCR products were routinely visualized by running 10 μl of PCR mixture on 1% agarose gels (Bio-Rad, Richmond, Calif.) in 1× Tris-acetate-EDTA (TAE) buffer stained with ethidium bromide (0.0001%). Reproducibility was confirmed by performing PCR with positive-control DNAs in triplicate as a minimum.
TABLE 2.
PCR primers and conditions for conventional PCR
| Primera | Target | Sequence | Tab (°C) | MgCl2 concn (mM) | Primer concn (μM) | Tpc (°C) | Expected product size (bp) |
|---|---|---|---|---|---|---|---|
| NAH-F | Naphthalene dioxygenase | 5′-CAAAA(A/G)CACCTGATT(C/T)ATGG | 47 | 2.5 | 0.3 | 83 | 377 |
| NAH-R | 5′-A(C/T)(A/G)CG(A/G)G(C/G)GACTTCTTTCAA | ||||||
| TOD-F | Toluene dioxygenase | 5′-ACCGATGA(A/G)GA(C/T)CTGTACC | 53 | 2.0 | 0.5 | 83 | 757 |
| TOD-R | 5′-CTTCGGTC(A/C)AGTAGCTGGTG | ||||||
| TOL-F | Xylene monooxygenase | 5′-TGAGGCTGAAACTTTACGTAGA | 55 | 2.5 | 0.2 | 82 | 475 |
| TOL-R | 5′-CTCACCTGGAGTTGCGTAC | ||||||
| BPH1-F | Biphenyl dioxygenase | 5′-GGACGTGATGCTCGA(C/T)CGC | 57 | 2.0 | 0.06 | 88 | 671 |
| BPH1-R | 5′-TGTT(C/G)GG(C/T)ACGTT(A/C)AGGCCCAT | ||||||
| BPH2-F | Biphenyl dioxygenase | 5′-GACGCCCGCCCCTATATGGA | 63 | 2.5 | 0.1 | 88 | 724 |
| BPH2-R | 5′-AGCCGACGTTGCCAGGAAAAT | ||||||
| BPH3-F | Biphenyl dioxygenase | 5′-CCGGGAGAACGGCAGGATC | 62 | 1.5 | 0.1 | 87 | 570 |
| BPH4-F | 5′-AAGGCCGGCGACTTCATGAC | 63 | 1.5 | 0.4 | 87 | 452 | |
| BPH3-Rd | 5′-TGCTCCGCTGCGAACTTCC | ||||||
| RMO-F | Toluene monooxygenase | 5′-TCTC(A/C/G)AGCAT(C/T)CAGAC(A/C/G)GACG | 53 | 3 | 0.4 | 82 | 466 |
| RMO-R | 5′-TT(G/T)TCGATGAT(C/G/T)AC(A/G)TCCCA | ||||||
| RDEG-F | Toluene monooxygenase | 5′-T(C/T)TC(A/C/G)AGCAT(A/C/T)CA(A/G)AC(A/C/G)GA(C/T)GA | 52 | 3 | 0.5 | 87 | 466 |
| RDEG-R | 5′-TT(A/G/T)TCG(A/G)T(A/G)AT(C/G/T)AC(A/G)TCCCA | ||||||
| PHE-F | Phenol monooxygenase | 5′-GTGCTGAC(C/G)AA(C/T)CTG(C/T)TGTTC | 49 | 4 | 0.3 | 86 | 206 |
| PHE-R | 5′-CGCCAGAACCA(C/T)TT(A/G)TC |
Forward (−F) and reverse (−R) primers are indicated.
Ta, the annealing temperature used during real-time PCR.
Tp, the polymerization temperature used during real-time PCR.
BPH3-R is used with BPH3-F and BPH4-F.
Hybridization experiments.
Hybridization studies with PCR products of each primer set with positive- and negative-control DNA were performed to confirm specificity. Probes were generated by PCR incorporation of digoxigenin-labeled dUTP. PCR products were separated on a 1% agarose gel and transferred to a nylon membrane (Hybond-N+; Amersham Pharmacia) by alkaline transfer. Hybridization was performed overnight at 25°C below the predicted melting temperature. Hybridization was performed under high (90%)- and medium (80%)-stringency conditions by adjusting the temperature of posthybridization washes (37). As noted in Results, low-stringency conditions (60%) were used as needed. Probes were detected according to the manufacturer's instructions (Roche Molecular Biochemicals, Indianapolis, Ind.). Prior to quantification by real-time PCR, conventional PCR products were prescreened by visualization on agarose gels to detect any nonspecific products and confirmed by hybridization.
Real-time PCR.
Real-time PCR was performed on an ABI 7700 Sequence Detector (Applied Biosystems, Foster City, Calif.). Q-PCR mixtures contained 1× cloned Pfu buffer (Stratagene, La Jolla, Calif.), 0.2 mM each of the four deoxynucleoside triphosphates, SYBR green (diluted 1:30,000; Molecular Probes, Eugene, Oreg.), and 1 U of PfuTurbo HotStart DNA polymerase (Stratagene). Annealing temperatures, primer concentrations, and MgCl2 concentrations for real-time PCR were identical to those for conventional PCR (Table 2). DNA standards ranging from 1 to 10−4 ng/μl were prepared from serial dilutions of DNA extracts from positive-control strains. To determine the melting temperatures of amplification products, melting curves were acquired by heating to 95°C for 1 min, cooling to 5°C below the annealing temperature, and ramping the temperature at 0.2°C/s to 95°C. Fluorescence measurements were taken during the final temperature ramp. The temperature of the extension step in subsequent PCRs was set at 4 to 5°C below the observed melting temperature. Melting curves were also routinely checked to confirm quantification of the desired product.
Threshold cycle number calculation.
The Sequence Detector program (version 1.7) subtracted background signal for each sample during cycles 3 through 15. The fluorescence threshold was computed as 10 times the standard deviation of the background signals, and fractional cycle numbers were computed and found to correlate inversely to the log of the initial template concentration. The best fit (by the least-squares method) was then used to plot the standard curve. The lower detection limit was defined as the lowest template concentration which resulted in a threshold cycle (Ct) that was significantly less than the total number of cycles performed (α = 0.05). Gene copy numbers were calculated from concentrations of positive-control standards assuming 9.12576 × 1014 bp/μg of DNA, 4.6 Mbp per genome, and one gene copy per genome.
RESULTS AND DISCUSSION
Phylogeny of the large subunits of aromatic oxygenases.
The large subunits of aromatic oxygenases with the same reported substrate specificity are, in general, closely related (Fig. 1), but distinct types are evident. The first type (N), consisting primarily of naphthalene dioxygenases, contains two families (N.1 and N.2) each with multiple subfamilies. Naphthalene dioxygenase-specific primers (NAH primers) target the N.2.A subfamily of naphthalene dioxygenases with high sequence identity to nahAc from Pseudomonas putida G7. The dntAc gene from Burkholderia sp. strain DNT belongs to this phylogenetic subfamily, as indicated by the relatively high DNA sequence identity to the naphthalene dioxygenase gene nagAc of Pseudomonas sp. strain U2 (92.1%). Furthermore, clones expressing dinitrotoluene dioxygenase convert naphthalene to the corresponding cis-dihydrodiol (49). The nahAc primers developed by Hamann et al. (18), Laurie and Lloyd-Jones (29), Wilson et al. (59), and in this study are based on reviews of a diverse group of nahAc genes and target the N.2.A subfamily (NAH primers). On the basis of our alignments, the 2-nitrotoluene dioxygenase primers (44) would amplify some but perhaps not all naphthalene dioxygenase genes from the N.2.A subfamily. The primary advantage of the NAH primers described here is that they target the entire N.2.A subfamily including dntAc and nagAc genes without mismatches. The other naphthalene dioxygenase subfamilies not targeted by the NAH primers are sequences from marine isolates (16, 19) and the polycyclic aromatic hydrocarbon-attacking dioxygenases from Alcaligenes faecalis AFK2 and Burkholderia sp. strain RP007. An additional nontarget sequence, narAa from Rhodococcus sp. strain NCIMB 12038, appears to be more closely related to biphenyl and toluene dioxygenases than other naphthalene dioxygenases (29).
FIG. 1.

(A) Phylogenetic analysis of the large subunits of aromatic dioxygenase genes. The tree was constructed by the neighbor-joining method (54) and bootstrapping analysis. Designations at branch points, e.g., D.1.A, indicate type (D), family (2), and subfamily (A). Subfamilies of genes were used to perform alignments leading to the identification of PCR primer sets. (B) Phylogenetic analysis of the large subunits of ring-hydroxylating monooxygenase genes. Isolates are described in Table 1.
The second type of aromatic dioxygenase is composed of two families of biphenyl and monoaromatic dioxygenases (Fig. 1, families D.1 and D.2). The sequence similarity and functional overlap of biphenyl and alkyl-benzene dioxygenases, including toluene dioxygenase, has been noted previously (14, 48). The D.1 family includes two subfamilies of biphenyl dioxygenases from gram-negative organisms (D.1.B and D.1.C) (50) and a subfamily of monoaromatic dioxygenase genes (D.1.A). The second family (D.2) is comprised of two subfamilies of biphenyl dioxygenases from gram-positive organisms (D.2.A and D.2.B) and a subfamily of toluene dioxygenases (D.2.C). The D.2.B subfamily included ipbA1 from R. erythropolis BD2 which had a higher DNA sequence identity to bphA1 from Rhodococcus sp. strain RHA1 than to isopropylbenzene dioxygenases from gram-negative organisms. Separate BPH primer sets were found to detect and distinguish between all four biphenyl dioxygenase subfamilies as shown in Fig. 1A. The BPH4 primers allow amplification of biphenyl and isopropylbenzene dioxygenases within the D.2 family, whereas the BPH3 primers are specific for the D.2.A subfamily. Biphenyl dioxygenase-specific primers have also been reported previously (44); however, sequence alignments with todC1 indicated only two mismatches, which suggests that toluene dioxygenase genes may also be amplified by this primer set. The D.2.C subfamily of dioxygenases for toluene, benzene, and chlorobenzene degradation, are closely related and were used to deduce toluene dioxygenase-specific primers (TOD primers).
The sequences of the large subunits of toluene monooxygenase genes were also aligned. The two types revealed with this alignment (Fig. 1B) differed in their mode of attack, ring-hydroxylating monooxygenases (type R) and alkyl group-hydroxylating monooxygenases (type T). With two exceptions, the ring-hydroxylating monooxygenases were divided into families on the basis of substrate specificity: two families of aromatic hydrocarbon-attacking monooxygenases (R.2 and R.3) and one family of phenol hydroxylases (R.1). The two exceptions are phlK from Ralstonia eutropha JMP134, which grouped with the toluene monooxygenases, and tbmD from Burkholderia sp. strain JS150, which is more closely related to the phenol hydroxylases. Johnson and Olsen (22) have previously shown that the tbm operon from strain JS150 has the same gene arrangement and strong sequence identity to the phenol hydroxylases encoded by the dmp, phe, and phh genes of strains CF600, BH, and P35X, respectively. Moreover, tbmD is also responsible for oxidation of o-cresol produced from the initial hydroxylation of toluene, supporting the association of this family with oxidation of hydroxylated substrates (23). Members of the R.2 and R.3 families will oxidize hydroxylated intermediates in addition to toluene (touA). PhlK has been described as a phenol hydroxylase, but its specificity has not been rigorously determined. Thus, it may also be active in toluene oxidation and belong to this phylogenetic family. On the basis of the apparent phylogeny, four primer sets were identified to detect each family as shown (Fig. 1B). RDEG primers were designed to amplify families R.2 and R.3, whereas the RMO primers are specific for the R.2 family. Species-specific primer sets which amplify fragments of todC1 from P. putida F1 and toluene-4-monooxygenase (tmoAa) from P. mendocina KR1 have been described in the literature (40, 44). However, these primers may not amplify benzene, toluene, and chlorobenzene dioxygenase genes related to todC1 or the R.2 family of toluene monooxygenases related to tmoA (Fig. 1B).
PCR primer testing and optimization.
To test specificity, PCR amplification with each primer set was performed with DNAs from positive-control strains containing the target and negative-control strains containing other oxygenase genes. For each primer set, amplification with positive-control DNA yielded amplification products of the predicted size. In most cases, no products were observed with negative-control DNA templates, but a few exceptions were noted. With reaction mixtures containing NAH primers and P. putida HS1 DNA as the template, an approximately 850-bp product which did not hybridize to the NAH/G7 probe was observed (Fig. 2). Amplification of a fragment of the toluene monooxygenase gene xylM with the NAH primers seems unlikely, since no products were observed when DNA from P. putida mt-2 was used. Although unexpected, this product was easily distinguished from the NAH product and did not generate false-positive results with environmental samples (B. R. Baldwin, C. H. Nakatsu, and L. Nies, unpublished data). Admittedly, the NAH primers described here and cited previously can be used to detect only a subset of naphthalene dioxygenase genes; however, detection of this subfamily may be an indicator of naphthalene catabolic ability (12, 17). PCR with the RMO primers produced an amplicon of approximately 460 bp with P. mendocina KR1 despite two predicted mismatches with each primer. The product weakly hybridized with the RMO probe constructed from JI104 template under low-stringency conditions. Thus, the RMO primers allowed amplification of ring-hydroxylating toluene monooxygenase genes from the R.2 and R.3 families. Amplicons characteristic of a phenol hydroxylase gene were observed in reactions with PHE primers and KR1 and JI104 DNAs. The product resulting from KR1 template hybridized under medium-stringency conditions to the PHE/CF600 probe, whereas the JI104 product hybridized to the probe only under low-stringency conditions. There should be sufficient mismatches between the PHE primers and the monooxygenase genes to prevent amplification; therefore, a gene downstream in the pathway was possibly amplified. Because both strains produce methyl-substituted phenols from toluene, downstream phenol hydroxylases would seem likely. A product characteristic of the BPH2 subfamily of biphenyl dioxygenases was also observed with JI104. Since the product hybridized to the BPH2/KF707 probe under high-stringency conditions and biphenyl will support its growth, JI104 is believed to contain a bph operon in addition to the known bmo operon.
FIG. 2.

NAH primer specificity confirmation by agarose gel electrophoresis. The specificity of each primer set was confirmed by PCR with positive- and negative-control templates (A), followed by hybridization experiments with gene probes created from positive-control strains (B). Lanes: M, 100- to 3,000-bp markers; 1, P. putida G7; 2, Burkholderia sp. strain DNT; 3, P. putida F1; 4, P. putida HS1; 5, P. putida mt-2.
Multiplex PCR.
Combinations of primer sets based on optimum annealing temperatures were tested in multiplex PCR to allow simultaneous detection and consequently, faster sample processing. Although the range of primer annealing temperatures excluded many combinations, the PHE/NAH, TOL/TOD, and BPH2/BPH4 primer sets shown in Fig. 3 allowed reliable detection in multiplex PCR experiments. Product yields with the TOL and TOD primers and the BPH2 and BPH4 primers were unchanged in multiplex reactions. Amplification of the PHE product was reduced slightly in multiplex reactions judging from product intensity, but products could still be observed with template concentrations of 0.1 ng per reaction mixture (2 × 104 copies per reaction mixture) in a 10-fold-excess P. putida G7 template. Additional combinations of multiplex reactions were attempted, but poor yield of one or more of the products would preclude their use for environmental samples.
FIG. 3.
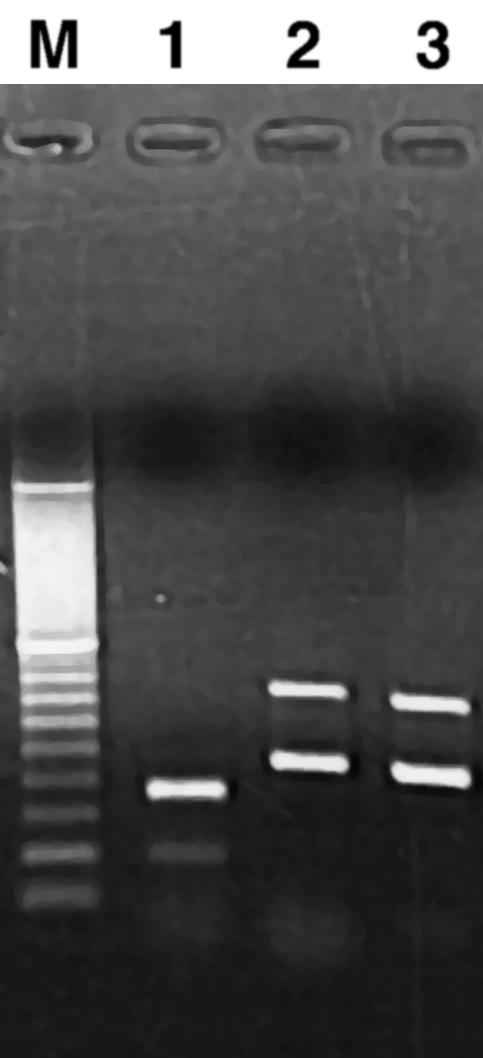
Multiplex PCR amplification. Multiplex PCR amplification performed with mixtures of positive-control DNAs. Lanes: M, 100- to 3,000-bp markers; 1, NAH/PHE primers with P. putida G7 and Pseudomonas sp. strain CF600 DNAs; 2, TOL/TOD primers with P. putida HS1 and P. putida F1 DNAs; 3, BPH2/BPH4 primers with P. pseudoalcaligenes KF707 and Rhodococcus sp. strain RHA1 DNAs.
An initial aromatic hydrocarbon catabolic screen consisting of PHE/NAH multiplex PCR, TOL/TOD multiplex PCR, and PCR with the RDEG primers could be used to document the presence of aromatic catabolic genotypes prior to quantification by real-time PCR. The biphenyl dioxygenase screen consisting of PCR with BPH1 primers and BPH2/BPH4 multiplex PCR would determine the presence of known biphenyl dioxygenase genes. Considering the cooccurrence of biphenyl and monoaromatic catabolic pathways demonstrated here by P. aeruginosa JI104, a biphenyl dioxygenase screening would be valuable for petroleum-contaminated sites in addition to sites in which polychlorinated biphenyls are encountered. Initial screening of environmental samples by multiplex PCR would reduce overall analysis cost, and any nonspecific products formed would be noted prior to real-time PCR.
Real-time PCR amplification.
To determine how easily real-time PCR could be conducted with existing primers, experiments were initially performed with the optimum conditions determined by conventional PCR. For some primer sets, no modifications of PCR conditions were needed and a log-linear relationship was observed between copy number and Ct. For others, a significant background fluorescence signal was observed even for no-template control samples. After determining that the signal in no-DNA controls was not a result of contamination, PfuTurbo HotStart DNA polymerase was used to decrease formation of any nonspecific products. This measure alone did not eliminate background signal, so melting curves were developed to aid in choosing an extension temperature (Fig. 4). The large change in fluorescence signal during the temperature ramp occurs at the melting point of the desired product (92°C). By collecting fluorescence data at extension temperatures near the melting temperature of the desired product, background fluorescence signals from primer dimers were greatly reduced. Temperatures of the extension step (Table 2) were as high as 88°C, which is considerably higher than the 72°C commonly used. Following optimization, standard curves were developed with known template concentrations. For all primer sets, a log-linear relationship was found between copy number and Ct values for template concentrations ranging from 2 × 106 to 2 × 102 copies per reaction mixture. Template concentrations greater than 2 × 106 copies inhibited amplification, as judged by increasing Ct values. The lower detection limit was 2 × 102 copies per reaction mixture for all primer sets. Reducing background fluorescence signal by using a HotStart polymerase and optimizing the polymerization temperature appear to be critical in decreasing detection limits.
FIG. 4.

(A) Melting curve of amplification product with primer BPH1. No-template control (○) and 1 ng of Rhodococcus sp. strain RHA1 (×) were used. The melting point of the PCR product can be identified by the peak in the plot of −dF/dT as a function of temperature during a 0.2°C/s temperature ramp at the end of the run. For the BPH4 product shown, the observed melting temperature is approximately 92°C. Subsequent real-time PCR with the BPH4 primers used a polymerization temperature of 87°C. (B) Amplification plot with primer BPH4. Different numbers of copies were used as follows: 107 (▪), 106 (□), 105 (⧫), 104 (◊), and 103 (•) copies per reaction mixture. ○, no-template control.
Isolation of aromatic hydrocarbon-degrading organisms has traditionally relied on selective enrichment, which has been shown to reduce diversity (9). Therefore, the phylograms shown in Fig. 1 represent the diversity only of currently known aromatic oxygenase gene sequences, not the true environmental diversity. As the number of available sequences increases, conserved regions used for primers can be identified more accurately and sequence variability can be assessed more thoroughly. This study was conducted to develop PCR primers and Q-PCR protocols to detect specific aromatic catabolic genotypes without excluding related but uncharacterized genes. Therefore, we reviewed available DNA sequences for conserved regions unique to each particular subfamily of oxygenase gene to use as PCR primers. In this manner, uncharacterized members of a subfamily are less likely to be excluded from detection.
At sites contaminated by gasoline and diesel fuel, aromatic hydrocarbons pose the greatest threat to human health and the environment. Consequently, concentrations of benzene, toluene, and xylenes (BTX) in groundwater are often measured quarterly and used as remediation endpoint criteria. Samples taken to document contaminant concentrations could also be readily available for DNA extractions and genetic characterization. In preliminary experiments with aquifer samples from gasoline-contaminated sites, oxygenase genes were routinely enumerated in contaminated wells, but none were detected in upgradient wells where BTX was not detected (B. R. Baldwin, L. Nies, and C. H. Nakatsu, submitted for publication). The real-time PCR assays described here can be used to enumerate catabolic genes involved specifically in the biodegradation of aromatic pollutants. Real-time PCR with oxygenase-specific primers will provide scientists and engineers with direct and more accurate feedback than culture-based assays to determine the effectiveness of monitored natural attenuation and to optimize operating variables (e.g., oxygen addition) and would complement contaminant removal data for site characterization. In addition, such a measure would allow investigation of the microbial ecology of petroleum-contaminated sites to determine the roles of currently known catabolic pathways in bioremediation in the field.
Acknowledgments
This work was supported in part by the Joint Transportation Research Project of the School of Civil Engineering at Purdue University, the Indiana Department of Transportation, the Purdue Research Foundation, and the Showalter Foundation.
We thank M. Slyvestre, E. Moore, K. Furukawa, G. S. Sayler, J. Spain, G. Zylstra, J. R. van der Meer, M. Roberts, D. Kunz, and P. Alaverez for providing strains used as controls.
REFERENCES
- 1.Aoki, H., T. Kimura, H. Habe, H. Yumane, T. Kodama, and T. Omori. 1996. Cloning, nucleotide sequence, and characterization of the genes encoding enzymes involved in the degradation of cumene to 2-hydroxy-6-oxo-7-methylocta-2,4-dienoic acid in Pseudomonas fluorescens IP01. J. Ferment. Bioeng. 81:187-196. [Google Scholar]
- 1a.Asturias, J. A., E. Dìaz, and K. N. Timmis. 1995. The evolutionary relationship of biphenyl dioxygenase from gram-positive Rhodococcus globerulus P6 to multicomponent dioxygenase from gram-negative bacteria. Gene 156:11-18. [DOI] [PubMed] [Google Scholar]
- 2.Becker, S., P. Boger, R. Oehlmann, and A. Ernst. 2000. PCR bias in ecological analysis: a case study for quantitative Taq nuclease assays in analyses of microbial communities. Appl. Environ. Microbiol. 66:4945-4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson, D. A., M. S. Boguski, D. J. Lipman, J. Ostell, B. F. Ouellette, B. A. Rapp, and D. L. Wheeler. 1999. GenBank. Nucleic Acids Res. 27:12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boronin, A. M., T. V. Tsoi, I. A. Kosheleva, M. U. Arinbasarov, and V. M. Adanin. 1989. Cloning of Pseudomonas putida genes responsible for the primary stages of oxidation of naphthalene in Escherichia coli cells. Genetika 25:226-237. [PubMed] [Google Scholar]
- 5.Bosch, R., E. Garcia-Valdes, and E. R. B. Moore. 1999. Genetic characterization and evolutionary implications of a chromosomally encoded naphthalene-degradation upper pathway from Pseudomonas stutzeri AN10. Gene 236:149-157. [DOI] [PubMed] [Google Scholar]
- 6.Byrne, A. M., J. J. Kukor, and R. H. Olsen. 1995. Sequence analysis of the gene cluster encoding toluene-3-monooxygenase from Pseudomonas pickettii PKO1. Gene 154:65-70. [DOI] [PubMed] [Google Scholar]
- 7.Corkery, D. M., and A. D. W. Dobson. 1998. Reverse transcription-PCR analysis of the regulation of ethylbenzene dioxygenase gene expression in Pseudomonas fluorescens CA-4. FEMS Microbiol. Lett. 166:171-176. [DOI] [PubMed] [Google Scholar]
- 8.Denome, S. A., D. C. Stanley, E. S. Olson, and K. D. Young. 1993. Metabolism of dibenzothiophene and naphthalene in Pseudomonas strains: complete DNA sequence of a upper naphthalene catabolic pathway. J. Bacteriol. 175:6890-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunbar, J., S. White, and L. J. Forney. 1997. Genetic diversity through the looking glass: effect of enrichment bias. Appl. Environ. Microbiol. 63:1326-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton, R. W., and K. N. Timmis. 1986. Characterization of a plasmid-specified pathway for catabolism of isopropylbenzene in Pseudomonas putida RE204. J. Bacteriol. 168:123-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erickson, B. D., and F. J. Mondello. 1992. Nucleotide sequencing and transcriptional mapping of genes encoding biphenyl dioxygenase, a multicomponent polychlorinated-biphenyl-degrading enzyme in Pseudomonas strain LB400. J. Bacteriol. 174:2903-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming, J. T., J. Sanseverino, and G. S. Sayler. 1993. Quantitative relationship between naphthalene catabolic gene frequency and expression in predicting PAH degradation in soils at town gas manufacturing sites. Environ. Sci. Technol. 27:1068-1074. [Google Scholar]
- 13.Fuenmayor, S. L., M. Wild, A. L. Boyes, and P. A. Williams. 1998. A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J. Bacteriol. 180:2522-2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furukawa, K., J. Hirose, A. Suyama, T. Zaiki, and S. Hayashida. 1993. Gene components responsible for discrete substrate specificity in the metabolism of biphenyl (bph operon) and toluene (tod operon). J. Bacteriol. 175:5224-5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furukawa, K., N. Hayase, K. Taira, and N. Tomizuka. 1989. Molecular relationship of chromosomal genes encoding biphenyl/polychlorinated biphenyl catabolism: some soil bacteria possess a highly conserved bph operon. J. Bacteriol. 171:5467-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiselbrecht, A. D., B. P. Hedlund, M. A. Tichi, and J. T. Staley. 1998. Isolation of marine polycyclic aromatic hydrocarbon (PAH)-degrading Cycloclasticus strains from the Gulf of Mexico and comparison of their PAH degradation ability with that of Puget Sound Cycloclasticus strains. Appl. Environ. Microbiol. 64:4703-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo, C., W. Sun, J. B. Harsh, and A. Ogram. 1997. Hybridization analysis of microbial DNA from fuel oil-contaminated and noncontaminated soil. Microb. Ecol. 34:178-187. [DOI] [PubMed] [Google Scholar]
- 18.Hamann, C., J. Hegemann, and A. Hildebrandt. 1999. Detection of polycyclic aromatic hydrocarbon degradation genes in different soil bacteria by polymerase chain reaction and DNA hybridization. FEMS Microbiol. Lett. 173:255-263. [DOI] [PubMed] [Google Scholar]
- 19.Hedlund, B. P., A. D. Geiselbrecht, T. J. Bair, and J. T. Staley. 1999. Polycyclic aromatic hydrocarbon degradation by a new marine bacterium, Neptunomonas naphthovorans gen. nov., sp. nov. Appl. Environ. Microbiol. 65:251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hino, S., K. Watanabe, and N. Takahashi. 1998. Phenol hydroxylase cloned from Ralstonia eutropha strain E2 exhibits novel kinetic properties. Microbiology 144:1765-1772. [DOI] [PubMed] [Google Scholar]
- 21.James, K. D., and P. A. Williams. 1998. ntn genes determining the early steps in the divergent catabolism of 4-nitrotoluene and toluene in Pseudomonas sp. strain TW3. J. Bacteriol. 180:2043-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson, G. R., and R. H. Olsen. 1995. Nucleotide sequence analysis of genes encoding a toluene/benzene-2-monooxygenase from Pseudomonas sp. strain JS150. Appl. Environ. Microbiol. 61:3336-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahng, H. Y., J. C. Malinverni, M. M. Majko, and J. J. Kukor. 2001. Genetic and functional analysis of the tbc operons for catabolism of alkyl- and chloroaromatic compounds in Burkholderia sp. strain JS150. Appl. Environ. Microbiol. 67:4805-4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kesseler, M., E. R. Dabbs, B. Averhoff, and G. Gottschalk. 1996. Studies on the isopropylbenzene 2,3-dioxygenase and the 3-iso-propylcatechol 2,3-dioxygenase genes encoded by the linear plasmid of Rhodococcus erythropolis BD2. Microbiology 142:3241-3251. [DOI] [PubMed] [Google Scholar]
- 25.Kimbara, K., T. Hashimoto, M. Fukuda, T. Koana, M. Takagi, M. Oishi, and K. Yano. 1989. Cloning and sequencing of two tandem genes involved in degradation of 2,3-dihydroxybiphenyl to benzoic acid in the polychlorinated biphenyl-degrading soil bacterium Pseudomonas sp. strain KKS102. J. Bacteriol. 171:2740-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosono, S., M. Maeda, F. Fuji, H. Arai, and T. Kudo. 1997. Three of the seven bphC genes of Rhodococcus erythropolis TA421, isolated from a termite ecosystem, are located on an indigenous plasmid associated with biphenyl degradation. Appl. Environ. Microbiol. 63:3282-3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurata, S., T. Kanagawa, K. Yamada, M. Torimura, T. Yokomaku, Y. Kamagata, and R. Kurane. 2001. Fluorescent quenching-based quantitative detection of specific DNA/RNA using a BODIPY®FL-labeled probe or primer. Nucleic Acids Res. 29:E34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin, M. J., C. C. R. Allen, L. A. Kulakov, and D. A. Lipscomb. 1999. Purification and characterization of a novel naphthalene dioxygenase from Rhodococcus sp. strain NCIMB12038. J. Bacteriol. 181:6200-6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laurie, A. D., and G. Lloyd-Jones. 1999. The phn genes of Burkholderia sp. strain RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J. Bacteriol. 181:531-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leander, M., T. Vallaeys, and R. Fulthorpe. 1998. Amplification of putative chlorocatechol dioxygenase gene fragments from α- and β-proteobacteria. Can. J. Microbiol. 44:482-486. [DOI] [PubMed] [Google Scholar]
- 30a.Ma, Y., and D. S. Herson. 2000. The catechol 2,3-dioxygenase gene and toluene monooxygenase genes from Burkholderia sp. AA1, an isolate capable of degrading aliphatic hydrocarbons and toluene. J. Ind. Microbiol. Biotechnol. 25:127-131. [Google Scholar]
- 31.Maeda, M., S.-Y. Chung, E. Song, and T. Kudo. 1995. Multiple genes encoding 2,3-dihydroxybiphenyl 1,2-dioxygenase in the gram-positive polychlorinated biphenyl-degrading bacterium Rhodococcus erythropolis TA421, isolated from a termite ecosystem. Appl. Environ. Microbiol. 61:549-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marmur, J. 1961. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3:209-218. [Google Scholar]
- 33.Masai, E., A. Yamada, J. M. Healy, T. Hatta, K. Kimbara, M. Fukuda, and K. Yano. 1995. Characterization of biphenyl catabolic genes of gram-positive polychlorinated biphenyl degrader Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 61:2079-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mesarch, M. B., and L. Nies. 1997. Modification of heterotrophic plate counts for assessing the bioremediation potential of petroleum-contaminated soil. Environ. Technol. 18:639-646. [Google Scholar]
- 35.Mesarch, M. B., C. H. Nakatsu, and L. Nies. 2000. Development of catechol 2,3-dioxygenase-specific primers for monitoring bioremediation by competitive quantitative PCR. Appl. Environ. Microbiol. 66:678-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison, T. B., J. J. Weis, and C. T. Wittwer. 1998. Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. BioTechniques 24:954-962. [PubMed] [Google Scholar]
- 37.Nakatsu, C. H., and L. J. Forney. 1996. Parameters of nucleic acid hybridization experiments, p. 1-12. In A. D. Akkermans, J. D. van Elsas, and F. J. de Bruijn (ed.), Molecular microbial ecology manual. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 38.Ng, L. C., V. Shingler, C. C. Sze, and C. L. Poh. 1994. Cloning and sequences of the first eight genes of the chromosomally encoded (methyl) phenol degradation pathway from Pseudomonas putida P35X. Gene 151:29-36. [DOI] [PubMed] [Google Scholar]
- 39.Nordlund, I., J. Powlowski, and V. Shingler. 1990. Complete nucleotide sequence and polypeptide analysis of multicomponent hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 172:6826-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogram, A., W. Sun, F. J. Brockman, and J. K. Fredrickson. 1995. Isolation and characterization of RNA from low-biomass deep-subsurface sediments. Appl. Environ. Microbiol. 61:763-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pflugmacher, U., B. Averhoff, and G. Gottschalk. 1996. Cloning, sequencing, and expression of isopropylbenzene degradation genes from Pseudomonas sp. strain JR1: identification of isopropylbenzene dioxygenase that mediates trichloroethene oxidation. Appl. Environ. Microbiol. 62:3967-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raeymaekers, L. 1993. Quantitative PCR: theoretical considerations with practical implications. Anal. Biochem. 214:582-585. [DOI] [PubMed] [Google Scholar]
- 43.Ridgway, H. F., J. Safarik, D. Phipps, P. Carl, and D. Clark. 1990. Identification and catabolic activity of well-derived gasoline-degrading bacteria from a contaminated aquifer. Appl. Environ. Microbiol. 56:3565-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ringelberg, D. B., J. W. Talley, E. J. Perkins, S. G. Tucker, R. G. Luthy, E. J. Bouwer, and H. L. Fredrickson. 2001. Succession of phenotypic, genotypic, and metabolic community characteristics during in vitro bioslurry treatment of polycyclic aromatic hydrocarbon-contaminated sediments. Appl. Environ. Microbiol. 67:1542-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Ecol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 46.Sayler, G. S., M. S. Shields, E. T. Tedford, A. Breen, S. W. Hooper, K. M. Sirotkin, and J. W. Davis. 1985. Application of DNA-DNA colony hybridization to the detection of catabolic genotypes in environmental samples. Appl. Environ. Microbiol. 49:1295-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon, M. J., T. D. Osslund, R. Saunders, B. D. Ensley, S. Suggs, A. A. Harcourt, W.-C. Suen, D. L. Cruden, D. T. Gibson, and G. J. Zylstra. 1993. Sequences of genes encoding naphthalene dioxygenase in Pseudomonas putida strain G7 and NCIB 9816-4. Gene 127:31-37. [DOI] [PubMed] [Google Scholar]
- 48.Smith, M. R., and C. Ratledge. 1989. Catabolism of biphenyl by Pseudomonas sp. NCIB 10643 and Nocardia sp. NCIB 10503. Appl. Microbiol. Biotechnol. 30:395-401. [Google Scholar]
- 49.Suen, W.-C., B. E. Haigler, and J. C. Spain. 1996. 2,4-Dinitrotoluene dioxygenase from Burkholderia sp. strain DNT: similarity to naphthalene dioxygenase. J. Bacteriol. 178:4926-4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sylvestre, M., M. Sirois, Y. Hurtubise, J. Bergeron, D. Ahmad, F. Shareck, D. Barriault, I. Guillemette, and J. M. Juteau. 1996. Sequencing of Comamonas testosteroni strain B-356-biphenyl/chlorobiphenyl dioxygenase genes: evolutionary relationships among gram-negative bacterial biphenyl dioxygenases. Gene 174:195-202. [DOI] [PubMed] [Google Scholar]
- 51.Taira, K., J. Hirose, S. Hayashida, and K. Furukawa. 1992. Analysis of bph operon from the polychlorinated biphenyl-degrading strain of Pseudomonas pseudoalcaligenes KF707. J. Biol. Chem. 267:4844-4853. [PubMed] [Google Scholar]
- 52.Takizawa, N., N. Kaida, S. Torigoe, T. Moritani, T. Sawada, S. Satoh, and H. Kiyohara. 1994. Identification and characterization of genes encoding polycyclic aromatic hydrocarbon dioxygenase and polycyclic aromatic hydrocarbon dihydrodiol dehydrogenase in Pseudomonas putida OUS82. J. Bacteriol. 176:2444-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan, H. M., H. Y. Tang, C. J. Joannou, N. H. Abdel-Wahab, and J. R. Mason. 1993. The Pseudomonas putida ML2 plasmid-encoded genes for benzene dioxygenase are unusual in codon usage and low in G+C content. Gene 130:33-39. [DOI] [PubMed] [Google Scholar]
- 54.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tyagi, S., and F. R. Kramer. 1996. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 14:303-308. [DOI] [PubMed] [Google Scholar]
- 56.Walia, S., A. Kahn, and N. Rosenthal. 1990. Construction and applications of DNA probes for detection of polychlorinated biphenyl-degrading genotypes in toxic organic contaminated soil environments. Appl. Environ. Microbiol. 56:254-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, Y., J. Garnon, D. Labbe, H. Bergeron, and P. C. K. Lau. 1995. Sequence and expression of the bpdC1C2BADE genes involved in the initial steps of biphenyl/chlorobiphenyl degradation by Rhodococcus sp. M5. Gene 164:117-122. [DOI] [PubMed] [Google Scholar]
- 58.Werlen, C., H. P. Kohler, and J. R. van der Meer. 1996. The broad substrate chlorobenzene dioxygenase and cis-chlorobenzene dihydrodiol dehydrogenase of Pseudomonas sp. strain P51 are linked evolutionarily to the enzymes for benzene and toluene degradation. J. Biol. Chem. 271:4009-4016. [DOI] [PubMed] [Google Scholar]
- 59.Wilson, M. S., C. Bakermans, and E. L. Madsen. 1999. In situ, real-time catabolic gene expression: extraction and characterization of naphthalene dioxygenase mRNA transcripts from groundwater. Appl. Environ. Microbiol. 65:80-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wittwer, C. T., M. G. Herrmann, A. A. Moss, and R. P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130-138. [DOI] [PubMed] [Google Scholar]
- 61.Yen, K.-M., and M. R. Karl. 1992. Identification of a new gene, tmoF, for the Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 174:7253-7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin, J. L., N. A. Shackel, A. Zekry, P. H. McGuinness, C. Richards, K. V. Putten, G. W. McCaughan, J. M. Eris, and G. A. Bishop. 2001. Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol. Cell Biol. 79:213-221. [DOI] [PubMed] [Google Scholar]
- 63.Zylstra, G. J., and D. T. Gibson. 1989. Toluene degradation by Pseudomonas putida F1: nucleotide sequence of the todC1C2BADE genes and their expression in E. coli. J. Biol. Chem. 264:14940-14946. [PubMed] [Google Scholar]