

Abstract
To determine the effects of glucocorticoids on the clearance of apoptotic and necrotic cells, the influence of dexamethasone on plasma levels of DNA was assessed in BALB/c mice receiving Jurkat cells treated with etoposide or ethanol. In untreated mice, administration of 108 apoptotic or necrotic Jurkat cells led to the appearance of DNA in the plasma. In mice treated 24 hours previously with dexamethasone, levels of DNA were reduced in a dose-dependent manner, with mice receiving 1 and 2.5 mg showing no appreciable plasma DNA levels. Similar results were obtained with assay of lactate dehydrogenase in mice receiving apoptotic cells. The effects of dexamethasone on anti-Fas treatment were also characterized. While treatment with a monoclonal anti-Fas reagent caused a significant plasma DNA response in untreated mice, mice pretreated with dexamethasone showed much lower levels. Blood levels of caspase 3 and TUNEL staining of liver were also reduced in dexamethasone-treated mice compared to controls receiving anti-Fas antibody. These results indicate that glucocorticoids can affect the clearance of apoptotic and necrotic cells as well as the induction of apoptosis in at least some tissues. These activities may be relevant to the efficacy of glucocorticoids in the treatment of inflammatory disease.
Cell death is an essential cellular process that occurs in both physiological and pathological settings. As defined by biochemical and morphological features, cells can die by two major pathways categorized as apoptosis and necrosis. Apoptosis or programmed cell death is a regulated process that is mediated by enzyme cascades that degrade key intracellular molecules. In contrast, necrosis or accidental cell death is a random process that results from physical or chemical events that cause irreparable cellular damage.1–3 While this dichotomy is an oversimplification, it provides a useful paradigm to investigate cell death.
The consequences of cell death extend beyond the loss of tissue. As shown in both in vivo and in vitro studies, dead cells can induce important immunological changes, with apoptotic cells showing anti-inflammatory activities and necrotic cells showing pro-inflammatory activities.4–6 These activities result at least in part from cellular macromolecules that can become either rearranged or released during the death process. Among these changes, the exposure of phosphatidylserine on the cell membrane during apoptosis can signal an anti-inflammatory state while release of the high mobility group-1 protein can cause a proinflammatory state.7–16 Other cellular molecules implicated in these activities include DNA, whose immunological activity may vary depending on concentration and protein binding.17–18
The extent to which dead cells expose or release internal molecules in an immunologically active form is determined by their uptake and clearance. These processes are highly efficient and result primarily by macrophages which bind dead and dying cells via cell surface molecules to promote phagocytosis.19–24 While the organism has a high capacity for clearance, the residue of dead and dying cells can nevertheless appear in the external milieu in certain circumstances as manifest, for example, by increased levels of circulating DNA in blood. High levels of DNA appear in diverse conditions such as systemic lupus erythematosus (SLE), cancer, and trauma and result from either an excessive burden of dead cells or impaired clearance.25–30
In recent studies, our laboratory has been investigating the clearance of dead and dying cells in the murine system to determine the circumstances in which DNA appears in the blood. As a model, we have assessed circulating levels of plasma DNA in mice that have received agents to induce in vivo apoptosis or have been infused with human Jurkat cells made apoptotic or necrotic in vitro.31,32 As shown previously, induction of in vivo apoptosis by LPS or anti-Fas antibody treatment, similar to administration of apoptotic and necrotic Jurkat cells, causes a prompt blood DNA response. In contrast, treatment with dexamethasone fails to induce elevation of blood DNA. These results are surprising, since dexamethasone causes significant thymocyte loss, raising the possibility that dexamethasone may affect events after the induction of cell death, including clearance.31,32
In the current experiments, we have addressed whether dexamethasone may modify the in vivo clearance of dead and dying cells in a way that affects DNA release into the external milieu. We have therefore measured levels of DNA in the blood in mice treated with dexamethasone and then infused with dead and dying cells or given anti-Fas to induce in vivo apoptosis.33,34 In results presented herein, we show that dexamethasone in a dose-dependent manner can prevent DNA release resulting from the administration of dead and dying cells as well as modify the consequences of anti-Fas treatment. Together, these findings indicate that glucocorticoids may modify events in the cell death process and provide evidence for a novel action of glucocorticoids that may be relevant to their use as anti-inflammatory agents to treat diseases such as SLE.
Materials and Methods
Administration of Cell Preparations
Female BALB/c mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and were used for experiments at ages 6 to 12 weeks. To assess the effects of glucocorticoids on DNA release, mice were pretreated with various doses of dexamethasone (Sigma Chemical Co, St. Louis, MO) 24 hours before the administration of apoptotic or necrotic cells or treatment with anti-Fas antibody. Both dexamethasone and anti-Fas were administered by the intraperitoneal (i.p.) route.
For the induction of apoptosis, Jurkat cells growing in RPMI 1640 with 10% fetal bovine serum (FBS) were treated with etoposide (Sigma Chemical Co.) at 30 μg/ml for 24 hours. For the induction of necrosis, cells were treated with 70% ethanol for 10 minutes. After treatment, cells were washed twice with phosphate-buffered saline (PBS) and suspended in PBS. The cells were administered i.p. at 108cells/mouse. Mice treated with anti-Fas antibody (product of BD PharMingen, San Diego, CA) received 10 μg/mouse. Following these treatments, mice were bled at regular times thereafter as indicated in the figures. The blood samples were collected into tubes with 3 to 5 μl of 0.5 mol/L EDTA, followed immediately by centrifugation. The plasma was removed and stored in −20°C until use.
Quantitation of Plasma DNA
Plasma DNA was assessed by a fluorimetric assay as previously described.35 Briefly, plasmas in various dilutions were mixed at a 1:1 ratio with the dye PicoGreen (Molecular Probes, Eugene, OR) diluted 1:200 in TE buffer (10 mmol/L Tris, 1 mmol/L EDTA, pH 8) in a black 96-well microtiter plate (Costar, Corning Incorporated, Corning, NY). The DNA concentration was determined from fluorescence measurements using a TECAN GENios microplate fluorescence reader (Salzburg, Austria), with an excitation wavelength at 485 nm and an emission wavelength at 535 nm. Data were collected as relative fluorescence units (RFU), with DNA concentrations in plasma were calculated according to a standard curve using double stranded calf thymus DNA (Sigma Chemical Co.)
Enzyme Assays
Lactate dehydrogenase (LDH) in plasma was measured with a cytotoxicity assay kit (Promega, Madison, WI). Briefly, plasma samples were diluted 1:500 in PBS/0.5% BSA and added to the substrate mix. After 30 minutes incubation at room temperature, the reaction was stopped and the concentration of LDH was determined by measuring the OD490 value. Data are reported as OD490 values. Caspase 3 was assayed using a caspase 3 kit (Molecular Probes). Briefly, plasma samples were diluted 1:100 in PBS and incubated with caspase 3 substrate at room temperature for 30 minutes. Fluorescence units were determined using a TECAN GENios microplate fluorescence reader as described above.
Flow Cytometry Analysis
To assess the effects of dexamethasone on immune cell populations, flow cytometry was performed on preparations of peritoneal washout cells and splenocytes. Briefly, mice were administered either 2.5 mg of dexamethasone/mouse in 100 μl of PBS or 100 μl of PBS alone as control. At 24 hours, mice were sacrificed by cervical dislocation, and peritoneal cells were harvested by lavage with 5 ml of cold RPMI 1640 medium (Gibco BRL, Grand Island, NY). Splenocytes were obtained by gently expressing the cells into a Petri dish containing cold RPMI 1640 medium using two microscope slides. The cells were pelleted at 300 × g for 5 minutes and resuspended in hypotonic lysis buffer to remove red blood cells, followed by centrifugation and two washes with PBS/0.5% BSA. The cell concentrations were adjusted to 1 × 107/ml in PBS. 100 μl (1 × 106 cells) were used for staining. Cells were treated with anti-mouse CD16/CD32 antibody (BD PharMingen), and then stained with either PE-anti-mouse F4/80 (Serotec, Raleigh, NC) or PE-anti-mouse Ly-6G, Ly-6C (BD PharMingen), double stained with annexin V-FITC (BD PharMingen). Cells were analyzed by FACScan flow cytometer (Becton Dickinson, Mansfield, MA). Data analysis was done using CellQuest software (Becton Dickinson Immunocytometry System, San Jose, CA).
Wright Staining
Peritoneal cells were harvested as described. Cytospin slides were stained with a Diff-Quik stain set (Dade Behring, Newark, DE).
TUNEL Staining
Livers were snap-frozen in Tissue Tek (Sakura Finetek, USA Inc. Torrance, CA) and 6-μm-thick sections were stained using a TdT FragEL kit (Oncogene Research Products, Boston, MA) for apoptotic cells. Cryosections were fixed in 4% formaldehyde and apoptotic nuclei were recognized by DNA fragment end-labeling, followed by streptavidin-peroxidase conjugate. DAB was used to detect the apoptotic cells and methyl green as counterstaining.
Statistical Analysis
Statistical analysis of the data were performed using Student’s two-tailed t-test. In figures and the table, values showing a statistically significant difference from controls at P < 0.05 are indicated by an asterisk.
Results
The initial experiments on the effects of dexamethasone used a model in which apoptotic or necrotic cells are administered to mice by the intraperitoneal route and plasma sampled thereafter for assessment of DNA by binding to PicoGreen. As shown previously, doses of 108 cells lead to a significant DNA response over the ensuing 24 hours, with apoptotic and necrotic cells producing similar findings.32 For the first experiments, mice were pretreated with 2.5 mg of dexamethasone 24 hours before administration of dead and dying cells. Pretreatment was performed after preliminary experiments showed greater consistency in the results than concomitant administration with dead cells. In addition, pretreatment may avoid potential confounding effects of simultaneous induction of apoptosis in thymocytes and other lymphoid populations.
Figure 1A shows time course experiments of plasma DNA levels in control mice and mice treated with dexamethasone at 2.5 mg and then administered 108 Jurkat cells treated with etoposide to induce apoptosis. As these results indicate, while normal mice receiving the apoptotic cells showed a rise in DNA levels over a 24-hour period, the mice receiving dexamethasone showed no increase in DNA levels over the same time period. Assays with LDH (Figure 1B) showed similar results.
Figure 1.

Effect of dexamethasone on plasma DNA levels from apoptotic cells. 108 Jurkat (JKT) cells, treated with etoposide to induce apoptosis, were injected i.p. into BALB/c mice (control or pretreated with 2.5 mg of dexamethasone 24 hours before). Levels of DNA (A) and LDH (B) were determined as described in Materials and Methods. Results are presented as means (±SD) for seven mice in each group. Values differing from control by P < 0.05 are indicated by an asterisk.
A dose-response experiment was next performed, testing doses of dexamethasone from 0.1 to 2.5 mg. Figure 2A shows that dexamethasone at doses of 1.0 and 2.5 mg markedly reduced the DNA response, while dexamethasone at 0.5 mg led to a delay in the peak response. As another measure of cellular death, LDH levels were measured. As shown in Figure 2B, while dexamethasone attenuated the LDH response at 2.5 mg, mice pretreated with dexamethasone at 1 mg still showed an increase.
Figure 2.

Dose response of the effects of dexamethasone. Dexamethasone at doses indicated was administered to BALB/c mice 24 hours before receiving 108 Jurkat cells treated with etoposide to induce apoptosis. DNA (A) and LDH (B) were measured as described. Results are reported as means (±SD) for four mice for controls and dexamethasone at 1 mg and 2.5 mg, and two mice for dexamethasone at 0.1 mg and 0.5 mg. Values differing from control by P < 0.05 are indicated by an asterisk.
The effects of administration of necrotic cells were tested next. Previous studies have indicated that administration of apoptotic and necrotic cells both lead to a rise in blood DNA, with similar time course and dose response.32 As shown in Figure 3, ethanol-treated Jurkat cells, like apoptotic Jurkat cells, led to a significant DNA response. As in the case of apoptotic cells, dexamethasone pretreatment prevented a rise in blood DNA, suggesting a more general effect on the handling of dead cells. With necrotic cells, however, there was no rise in LDH levels, likely reflecting the denaturation of the enzyme by ethanol treatment.
Figure 3.

Effect of dexamethasone on plasma DNA levels from necrotic cells. 108 Jurkat cells treated with ethanol (Etoh) to induce necrosis were administered to BALB/c mice and plasma levels of DNA and LDH determined. Results are reported as means (±SD) for six or seven mice. Values differing from control by P < 0.05 are indicated by an asterisk.
To investigate possible mechanisms for the effects of dexamethasone, cell populations in the peritoneum and spleen were analyzed by flow cytometry. As shown in Table 1, treatment with dexamethasone led to a significant increase at 24 hours in the number of neutrophils among peritoneal washout cells as well as spleen cells as measured by Gr-1 staining by flow cytometry. This finding was corroborated by Wright staining of peritoneal washouts showing an increase in the number of neutrophils (Figure 4). Although the number of annexin-positive neutrophils was not increased among peritoneal washout cells, the spleen showed an increase in the number of both annexin-positive, Gr-1-positive and annexin-positive, F4/80-positive cells. These results indicate that, at the time dead and dying cells were administered to mice in these experiments, dexamethasone had caused a significant alteration in the cellular composition of various lymphoid compartments, including a major increase in the number of neutrophils as well as an increased frequency of apoptotic neutrophils and macrophages in the spleen.
Table 1.
Flow Cytometry Analysis of Cell Populations
| Peritoneum (%)
|
Spleen (%)
|
|||
|---|---|---|---|---|
| PBS | Dexamethasone | PBS | Dexamethasone | |
| Gr-1 (+) | 10.70 (±1.88) | 43.21 (±5.15)* | 5.31 (±2.25) | 22.23 (±7.90)* |
| Gr-1(+) annexin V(+) | 18.49 (±1.62) | 18.13 (±2.25) | 1.06 (±0.21) | 2.97 (±0.59)* |
| F4/80(+) | 35.86 (±2.65) | 28.82 (±3.17) | 3.20 (±0.23) | 3.28 (±0.19) |
| F4/80(+) annexin V(+) | 2.49 (±0.51) | 1.56 (±0.46) | 1.68 (±0.38) | 4.40 (±0.41)* |
Cells were analyzed by flow cytometry with staining by Gr-1 and annexin V or F4/80 and annexin V to assess cell populations arising 24 hours after treatment with either PBS or dexamethasone. Results represent mean ± SEM of % cells, with N = 9–14.
Student’s t-test, P < 0.05.
Figure 4.

Effect of dexamethasone on peritoneal cell population. Wright staining was performed on Cytospin slides of peritoneal washout cells from normal BALB/c mice (A) and mice receiving 100 μl of PBS (B) or 2.5 mg of dexamethasone in 100 μl of PBS (C). Images were taken at ×600 magnification.
In these experiments, the source of the dead and dying cells was exogenous. To determine whether dexamethasone would also affect an endogenous source of dead cells, the effects of dexamethasone on anti-Fas was next assessed. Treatment with a monoclonal anti-Fas antibody Jo2 causes massive hepatic cells death as well as death of lymphoid cell populations which, as shown in previous experiments, lead to high blood DNA levels before animal death.32–34
Figure 5 shows results of DNA measurements of plasma of control and dexamethasone treated mice receiving 10 μg of anti-Fas. As these data indicate, treatment of mice with dexamethasone prevents the DNA increase resulting from anti-Fas treatment. Similar results were obtained with the LDH assay. Since control mice treated with Jo2 died with this treatment, only the results of the 8-hour time point are shown. As these data indicate, dexamethasone prevents the rise in both DNA and LDH levels following anti-Fas treatment and, in addition, increases animal survival, with all six mice surviving beyond 48 hours, the point of the termination of the experiment.
Figure 5.

Effect of dexamethasone on plasma DNA levels from anti-Fas treatment. BALB/c mice (control or pretreated with 2.5 mg of dexamethasone) were given 10 μg of a monoclonal anti-Fas antibody (Jo2) and plasma obtained for determination of DNA (A), LDH (B), or caspase 3 (C). Results are reported as means (±SD) of six mice. Values differing from control by P < 0.05 are indicated by an asterisk.
Since dexamethasone prevents release of DNA as well as LDH, these findings suggest that this treatment could affect the occurrence of apoptosis as well as the clearance of dead cells. As an approach to this issue, levels of caspase 3 in the blood were assessed. As demonstrated by Hentze et al,36 levels of this enzyme, which is released by apoptotic cells, correlates with apoptosis both in vitro and in vivo. As shown in Figure 5, caspase 3 levels increased dramatically after anti-Fas treatment whereas, after treatment with dexamethasone, levels of this enzyme did not rise after anti-Fas treatment. These results suggest that hepatocyte apoptosis was prevented. This possibility is supported by TUNEL analysis of liver sections (Figure 6), showing reduced TUNEL staining in livers of mice treated with dexamethasone before receiving anti-Fas antibody. Taken together with results on DNA and LDH after anti-Fas treatment, these suggest that dexamethasone may affect the occurrence of apoptosis in certain sites as well as the clearance of apoptotic cells.
Figure 6.
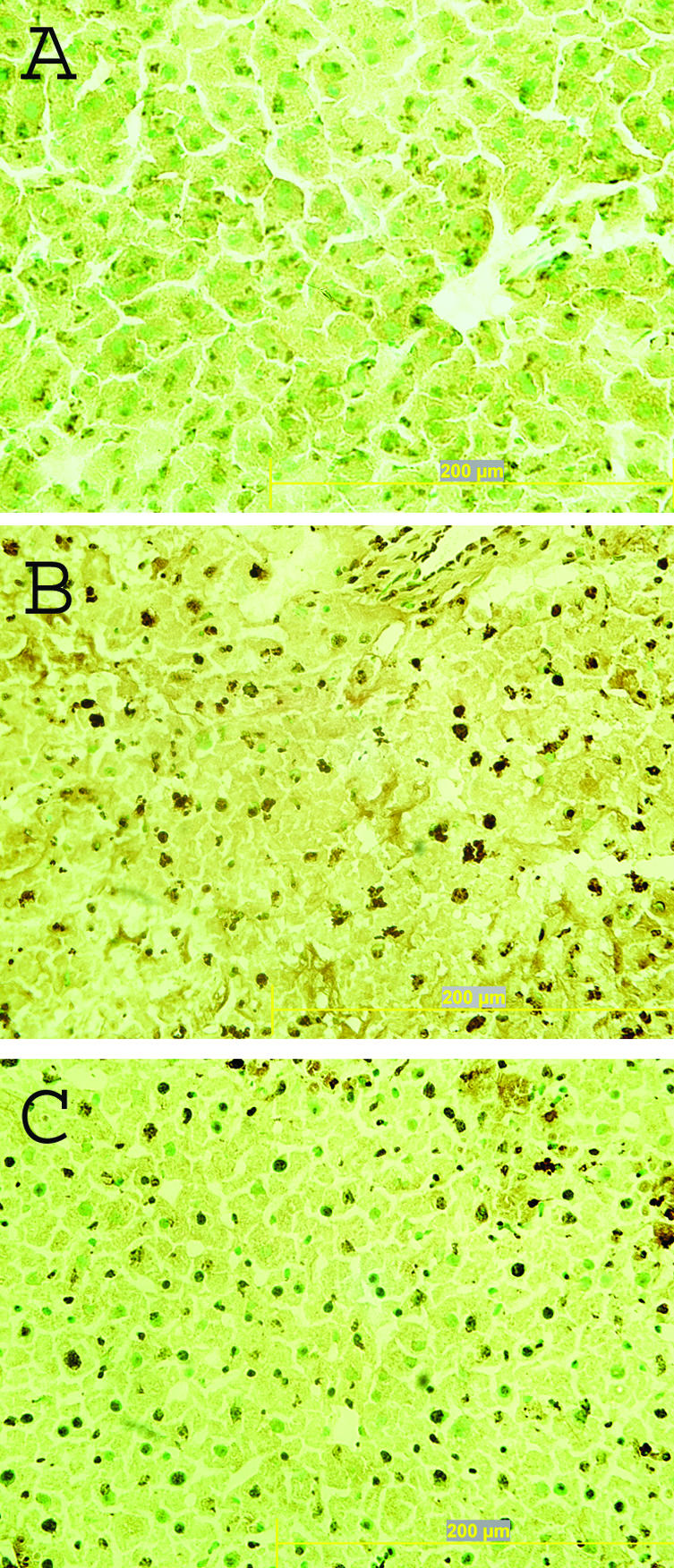
Effect of dexamethasone on hepatocytes from anti-Fas treatment. TUNEL staining was performed on cryosections of livers from normal BALB/c mice (A) and mice receiving 10 μg of anti-Fas antibody without (B) or with (C) dexamethasone pretreatment. Apoptotic cells are stained brown and normal cells are shown by methyl green counterstaining. Images were taken at ×400 magnification.
Discussion
Results present herein provide new insights into the pathways for the clearance of apoptotic and necrotic cells and the manner in which DNA resulting from cell death enters the circulation. Thus, our data indicate that dexamethasone can inhibit in a dose-dependent fashion the plasma DNA response following either administration of dead or dying cells or the in vivo induction of apoptosis by anti-Fas. Since this treatment reduces the release of LDH as well as DNA, the effect of dexamethasone appears to involve more than one macromolecule, suggesting an effect on the clearance of the cells themselves as opposed to the metabolism of only DNA. Taken together with prior studies on the effects of glucocorticoids on the uptake of apoptotic cells,37–39 these findings suggest that glucocorticoids may play an important role in modifying not only the process of apoptosis but the consequences of exposure to cellular contents.
In these studies, we have used the administration of apoptotic and necrotic Jurkat cells as a model to investigate the clearance process. As shown previously, intraperitoneal administration of apoptotic and necrotic cells leads in a dose-dependent way to the appearance of DNA as well as nucleosomes in the blood.32 The source of this DNA can be readily determined since Jurkat cells are from a human male, allowing the use of a Y chromosomal sequence as a marker. As shown by PCR analysis, the DNA in the blood after administration of Jurkat cells arises primarily from the human cell line although DNA from the murine host, as measured using a GAPDH sequence, also contributes. Furthermore, plasma DNA that appears after administration of apoptotic and necrotic cells shows laddering by gel electrophoresis, implying nuclease digestion consistent with apoptosis.32
In previous experiments on the origin of circulating DNA, we assessed the responses of mice in which macrophages were eliminated by treatment with clodronate liposomes. This treatment, which causes macrophage death by apoptosis,40–42 leads to an initial rise in blood DNA, likely reflecting the generation of apoptotic cells and the absence of a clearance system. Subsequent administration of apoptotic or necrotic cells, however, fails to produce a blood DNA response. Silica treatment, which can functionally inactivate macrophages,43,44 also prevents the blood DNA response that occurs after administration of dead and dying cells; in our studies, however, silica did not cause an initial DNA elevation. These observations suggest that a blood DNA response involves a role of macrophages and does not simply reflect the disintegration of apoptotic or necrotic cells.
Similar to the administration of apoptotic and necrotic cells, in vivo induction of apoptosis can lead to a blood DNA response depending on the inducing agent,. Thus, treatment of normal mice with LPS and anti-Fas, but not dexamethasone, can cause significant blood DNA responses.31 Since these agents all induce in vivo apoptosis, these findings suggest that the appearance of DNA after in vivo cell death may depend on the site of cell death (eg, liver or thymus), the cell type killed (eg, hepatocyte or lymphocyte) as well as effects of the inducing agent on the clearance process. For example, LPS can induce cytokines that modulate macrophage function while anti-Fas may affect the viability or function of macrophages expressing Fas.45 The difference in the levels of DNA following in vivo apoptosis may also depend on the number of cells undergoing apoptosis, although assessing the total cells killed is difficult because of uncertainty in their locale and their transience following apoptosis.
The lack of a blood DNA response following administration of dexamethasone is striking in view of the large number of thymocytes killed.31 While there are many explanations for this finding, we were interested in the possibility that glucocorticoids can modulate either the clearance of apoptotic cells or their subsequent metabolism to generate circulating DNA. As shown by other investigators, glucocorticoids can affect the uptake of apoptotic cells by macrophages in vitro although these prior experiments did not measure the release of DNA from cells.37–39 Our data extend these findings and demonstrate clearly that glucocorticoids have an important influence on the generation of DNA into the blood. The effect was manifest with both apoptotic and necrotic cell administration as well as treatment of mice with an anti-Fas antibody.
The effects of dexamethasone resemble those induced by both clodronate and silica, agents which either eliminate or inactivate macrophage. In mice treated with these agents, administration of apoptotic or necrotic cells fails to produce a blood DNA response under conditions in which normal mice show high levels of DNA for as long as 24 hours.32 As discussed previously, this effect could result from a failure of macrophages to take up the dead cell which gradually disintegrate or break apart in a manner that does raise blood DNA levels. While glucocorticoids do not cause generalized macrophage apoptosis, they may cause selective loss of macrophage populations.39,46–48 It is possible, therefore, that the in vivo administration of dexamethasone reduces a cell population critical to the clearance of dead and dying cells. In the absence of this cell population, the uptake process may be prevented, limiting DNA release.
In our experiments, mice treated with dexamethasone showed a significant alteration in the cell composition of peritoneum and spleen, with both compartments showing a dramatic increase in neutrophil numbers. This effect can be attributed to the anti-apoptotic activity of glucocorticoids on neutrophils which are short-lived cell programmed to die. The prolongation of neutrophil lifespan, which can be augmented by various mediators and cytokines, may be important in host defense by increasing the number of phagocytic cells as well as during the resolution of inflammation by preventing the breakdown of neutrophils with the resultant spillage of their toxic contents.39,49–51 As suggested by our findings as well as others, glucocorticoids produce a situation in which large numbers of neutrophils, the usual feature of exudates, exist in a microenvironment in which other aspects of the inflammatory process may be blunted or blocked.
While glucocorticoids can increase the life span of neutrophils, these cells must eventually die at which time they may induce other changes encountered in inflammatory reactions. These events may include macrophage apoptosis. Indeed, in our experiments, the spleen showed an increase in the frequency of annexin-positive, F4/80-positive cells, suggesting that, in our model, macrophages may be undergoing apoptosis. In this regard, studies on the carageenan-induced pleurisy model indicate that macrophage apoptosis occurs during resolution of inflammation and follows neutrophil apoptosis.52
Together, these findings thus raise the possibility that macrophage dysfunction or loss may occur during the clearance of large numbers of neutrophils, whether arising during inflammation or secondary to glucocorticoid action. Furthermore, since glucocorticoids directly induce apoptosis in eosinophils, the effects of dead and dying cells on macrophages could be amplified and prolonged in time.50 While these considerations are speculative, they nevertheless suggest the possibility that the effects of glucocorticoids on macrophage number and function may be indirect and result from downstream effects in other cell populations.
In our previous studies, we considered the possibility that blood DNA occurs only when the normal clearance process is overwhelmed.32 At least two possibilities can be proposed for the consequence of overwhelming clearance. Thus, when the macrophage has engulfed excessive cells, DNA may be discharged from macrophages in an incompletely digested form that retains duplex structure. A second possibility concerns the toxicity of dead cells for macrophages. According to this scenario, macrophages, during phagocytosis of high numbers of dead cells, could undergo apoptosis, causing release of its own DNA and that of the engulfed dead cells. The finding of both murine and human DNA in the blood after administration of dead cells to normal mice is consistent with this possibility.32 Furthermore, as discussed above, the effects of glucocorticoids on neutrophils and eosinophils could eventuate in a similar situation.
An alternative possibility to explain the effects of dexamethasone on the blood DNA response concerns a glucocorticoid-induced enhancement of the clearance process. Among its activities in this system, dexamethasone may increase the efficiency of macrophage clearance of dead cells or make them resistant to the apoptosis that occurs with excessive phagocytosis of dead cells. With the macrophage bolstered by glucocorticoids, the clearance process would be enhanced, limiting release of DNA into the external milieu. The effect is both more simple and direct than that postulated above. In studies thus far in vivo (N. Jiang and D. Pisetsky, unpublished observations), however, we have not yet been able to demonstrate increased uptake or clearance of dead cells by macrophage. Since the utilization of large numbers of dead cells in our model limits enumeration of endogenous cell populations, studies are in progress to explore models where the number of administered dead cells is lower.
While the explanation for the activities of dexamethasone on the clearance of exogenous cells awaits further investigation, it is of interest that dexamethasone affects DNA release in the anti-Fas system as well as the Jurkat system despite the important differences in these models. Anti-Fas agent causes massive liver cell death and mortality, with apoptosis induced in hepatocytes by direct engagement of Fas.33,34 Nevertheless, in our experiments, mice pretreated with dexamethasone showed minimal release of DNA, LDH and caspase 3 as well as improved survival. These findings suggest either a direct protective effect of dexamethasone on the hepatocyte or an indirect effect by modulating the activity of macrophages (or other inflammatory cells) in the liver. The latter possibility is supported by findings in other systems on the effect of cytokines (eg, IL-6, TNF) on induction of apoptosis by anti-Fas as well as hepatic toxins. In these systems, modulation of cytokine levels, with consequent effects on anti-apoptotic proteins, modify the occurrence of cell death.53–56 The absence of a rise of caspase 3 after anti-Fas in dexamethasone-treated mice is consistent with prevention of apoptosis.
As these considerations indicate, glucocorticoids may have diverse effects on both apoptosis itself as well as subsequent events in the clearance of dead and dying cells. Since dead and dying cells can have important immunological effects, glucocorticoids have the potential to alter the immune environments in settings (eg, SLE, sepsis, toxin exposure) characterized by high burdens of apoptotic cells. In the setting of SLE, the effects of glucocorticoids on DNA release may be particularly important since DNA in the circulation can serve as a driving antigen for anti-DNA production as well as form immune complexes that can both stimulate cytokine production and deposit in the tissue.17,18 Studies are in progress to characterize further the effects of glucocorticoids on DNA release into the circulation in various pathological settings.
Footnotes
Address reprint requests to Dr. D. Pisetsky, Durham VA Medical Center, Box 151G, 508 Fulton St., Durham, NC 27705. E-mail: dpiset@acpub.duke.edu.
Supported by a VA Merit Review grant, an Alliance for Lupus Research grant, and National Institutes of Health grant AI44808.
References
- Farber E. Ideas in pathology: programmed cell death: necrosis versus apoptosis. Mod Pathol. 1994;7:605–609. [PubMed] [Google Scholar]
- Majno G, Joris I. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Leist M, Nicotera P. Breakthroughs and views: the shape of cell death. Biochem Biophys Res Commun. 1997;236:1–9. doi: 10.1006/bbrc.1997.6890. [DOI] [PubMed] [Google Scholar]
- McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-β in macrophages that have ingested apoptotic cells. J Immunol. 1999;163:6164–6172. [PubMed] [Google Scholar]
- Ronchetti A, Rovere P, Iezzi G, Galati G, Heltai S, Protti MP, Garancini MP, Manfredi AA, Rugarli C, Bellone M. Immunogenicity of apoptotic cells in vivo: role of antigen load, antigen-presenting cells, and cytokines. J Immunol. 1999;163:130–136. [PubMed] [Google Scholar]
- Reddy SM, Hsiao K-H K, Abernethy VE, Fan H, Longacre A, Lieberthal W, Rauch J, Koh JS, Levine JS. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J Immunol. 2002;169:702–713. doi: 10.4049/jimmunol.169.2.702. [DOI] [PubMed] [Google Scholar]
- Henson PM, Bratton DL, Fadok VA. The phosphatidylserine receptor: a crucial molecular switch? Nature Rev. 2001;2:627–633. doi: 10.1038/35085094. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166:6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. 1998;191:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Carpio DF, Zheng Y, Bruzzzo P, Singh V, Ouaaz F, Medzhitov RM, Beg AA. An essential role of the NF-κB/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- Verhoven B, Krahling S, Schlegel RA, Williamson P. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ. 1999;6:262–270. doi: 10.1038/sj.cdd.4400491. [DOI] [PubMed] [Google Scholar]
- Rendon-Mitchel B, Ochani M, Li J, Han J, Wang H, Yang H, Susarla S, Czura C, Mitchel RA, Chen G, Sama AE, Tracey KJ, Wang H. IFN-γ induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–3897. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- Andersson BU, Wang H, Palmblad K, Aveberger A-C, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallin H, Perers A, Alm GV, Ronnblom L. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol. 1999;163:6306–6313. [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:595–598. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ. 1998;5:563–568. doi: 10.1038/sj.cdd.4400407. [DOI] [PubMed] [Google Scholar]
- Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- Platt N, da Silva RP, Gordon S. Recognizing death: the phagocytosis of apoptotic cells. Trends Cell Biol. 1988;8:365–372. doi: 10.1016/s0962-8924(98)01329-4. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Odaka C, Mizuochi T. Macrophages are involved in DNA degradation of apoptotic cells in murine thymus after administration of hydrocortisone. Cell Death Differ. 2002;9:104–112. doi: 10.1038/sj.cdd.4400941. [DOI] [PubMed] [Google Scholar]
- Steinman CR. Circulating DNA in systemic lupus erythematosus: isolation and characterization. J Clin Invest. 1984:832–841. doi: 10.1172/JCI111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YMD, Chan LYS, Chan ATC, Leung S-F, Lo K-W, Zhang J, Lee JCK, Hjelm NM, Johnson PJ, Huang DP. Quantitative and temporal correlation between circulating cell-free Epstein-Barr virus DNA and tumor recurrence in nasopharyngeal carcinoma. Cancer Res. 1999;59:5452–5455. [PubMed] [Google Scholar]
- Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch R-D, Knippers R. DNA Fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- Rainer TH. Plasma DNA, prediction and post-traumatic complications. Clin Chim Acta. 2001;313:81–85. doi: 10.1016/s0009-8981(01)00653-2. [DOI] [PubMed] [Google Scholar]
- Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001;313:139–142. doi: 10.1016/s0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- Licht R, van Bruggen MCJ, Oppers-Walgreen B, Rijki TPM, Berden JHM. Plasma levels of nucleosomes and nucleosome-autoantibody complexes in murine lupus: effects of disease progression and lipopolysaccharide administration. Arthritis Rheum. 2001;44:1320–1330. doi: 10.1002/1529-0131(200106)44:6<1320::AID-ART224>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Jiang N, Reich CF, Monestier M, Pisetsky DS. The expression of plasma nucleosomes in mice undergoing in vivo apoptosis. Clin Immunol. 2003;106:139–147. doi: 10.1016/s1521-6616(02)00027-x. [DOI] [PubMed] [Google Scholar]
- Jiang N, Reich CF, Pisetsky DS. The role of macrophages in generation of circulating blood nucleosomes from dead and dying cells. Blood. 2003;102:2243–2250. doi: 10.1182/blood-2002-10-3312. [DOI] [PubMed] [Google Scholar]
- Ogasawara J, Watanabe-Fukunagea R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, Runkel L. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–1230. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman L, Reich CF, Pisetsky DS. The use of fluorometric assays to assess the immune response to DNA in murine systemic lupus erythematosus. Scand J Immunol. 2003;57:525–533. doi: 10.1046/j.1365-3083.2003.01261.x. [DOI] [PubMed] [Google Scholar]
- Hentze H, Schwoebel F, Lund S, Kehl M, Ertel W, Wendel A, Jäättelä M, Leist M. In vivo and in vitro evidence for extracellular caspase activity released from apoptotic cells. Biochem Biophys Res Commun. 2001;283:1111–1117. doi: 10.1006/bbrc.2001.4918. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, Dransfield I, Savill J, Rossi AG. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162:3639–3646. [PubMed] [Google Scholar]
- Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol. 2001;167:976–986. doi: 10.4049/jimmunol.167.2.976. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol. 2003;178:29–36. doi: 10.1677/joe.0.1780029. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A, van den Berg TK. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1996;193:93–99. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Selander KS, Mönkkönen J, Karhukorpi E-K, Härkönen P, Hannuniemi R, Väänänen HK. Characteristics of clodronate-induced apoptosis in osteoclasts and macrophages. Mol Pharmacol. 1996;50:1127–1138. [PubMed] [Google Scholar]
- Iyer R, Hamilton RF, Li L, Holian A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol Appl Pharmacol. 1996;141:84–92. doi: 10.1006/taap.1996.0263. [DOI] [PubMed] [Google Scholar]
- Iyer R, Holian A. Involvement of the ICE family of proteases in silica-induced apoptosis in human alveolar macrophages. Am J Physiol. 1997;273:L760–L767. doi: 10.1152/ajplung.1997.273.4.L760. [DOI] [PubMed] [Google Scholar]
- Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, Liles WC. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med. 1997;185:1511–1516. doi: 10.1084/jem.185.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci. 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Pauels H-G, Lügering N, Lügering A, Domschke W, Kucharzik T. Glucocorticoids induce apoptosis in human monocytes: potential role of IL-1β. J Immunol. 1999;163:3484–3490. [PubMed] [Google Scholar]
- Fingerle-Rowson G, Angstwurm M, Andreesen R, Zieler-Heitbrock HWL. Selective depletion of CD14+ CD16+ monocytes by glucocorticoid therapy. Clin Exp Immunol. 1998;112:501–506. doi: 10.1046/j.1365-2249.1998.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils: separation of survival and activation outcomes. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- Nittoh T, Fujimori H, Kozumi Y, Ishihara K, Mue S, Ohuchi K. Effects of glucocorticoids on apoptosis of infiltrated eosinophils and neutrophils in rats. Eur J Pharmacol. 1998;354:73–81. doi: 10.1016/s0014-2999(98)00426-9. [DOI] [PubMed] [Google Scholar]
- Ward C, Dransfield I, Chilvers ER, Haslett C, Rossi AG. Pharmacological manipulation of granulocyte apoptosis: potential therapeutic targets. Trends Pharmacol Sci. 1999;20:503–509. doi: 10.1016/s0165-6147(99)01391-7. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Colville-Nash PR, McMaster S, Sawatzky DA, Willoughby DA, Lawrence T. Inducible cyclooxygenase-derived 15-deoxy(delta)12–14PGJ2 brings about acute inflammatory resolution in rat pleurisy by inducing neutrophil and macrophage apoptosis. FASEB J. 2003;17:2269–2271. doi: 10.1096/fj.02-1162fje. [DOI] [PubMed] [Google Scholar]
- Küsters S, Gantner F, Künstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111:462–471. doi: 10.1053/gast.1996.v111.pm8690213. [DOI] [PubMed] [Google Scholar]
- Ksontini R, Colagiovanni DB, Josephs MD, Edward CK, III, Tannahill CL, Solorzano CC, Norman J, Denham W, Clare-Salzier M, MacKay SLD, Moldawer LL. Disparate roles for TNF-α and Fas Ligand in concanavalin A-induced hepatitis. J Immunol. 1998;160:4082–4089. [PubMed] [Google Scholar]
- Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev. 2000;174:160–171. doi: 10.1034/j.1600-0528.2002.017411.x. [DOI] [PubMed] [Google Scholar]
- Kovalovich K, Li W, DeAngelis R, Greenbaum LE, Ciliberto G, Taub R. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem. 2001;276:26605–26610. doi: 10.1074/jbc.M100740200. [DOI] [PubMed] [Google Scholar]