

Abstract
Development of diabetes generally reflects an inadequate mass of insulin-producing β-cells. β-cell proliferation and differentiation are regulated by a variety of growth factors and hormones, including insulin-like growth factor I (IGF-I). GRF1 is a Ras-guanine nucleotide exchange factor known previously for its restricted expression in brain and its role in learning and memory. Here we demonstrate that GRF1 is also expressed in pancreatic islets. Interest ingly, our GRF1-deficient mice exhibit reduced body weight, hypoinsulinemia and glucose intolerance owing to a reduction of β-cells. Whereas insulin resistance is not detected in peripheral tissues, GRF1 knockout mice are leaner due to increased lipid catabolism. The reduction in circulating insulin does not reflect defective glucose sensing or insulin production but results from impaired β-cell proliferation and reduced neogenesis. IGF-I treatment of isolated islets from GRF1 knockouts fails to activate critical downstream signals such as Akt and Erk. The observed phenotype is similar to manifestations of preclinical type 2 diabetes. Thus, our observations demonstrate a novel and specific role for Ras-GRF1 pathways in the development and maintenance of normal β-cell number and function.
Keywords: cell growth/β-cells/differentiation/insulin/Ras-GRF1
Introduction
Ras proteins are able to regulate various signaling pathways controlling cell growth, differentiation or survival. They do so through their ability to integrate multiple, different upstream signals and to elicit a variety of different cellular responses. The broad array of signals emanating from Ras reflects the binding and activation of different downstream effectors. Known effectors of Ras signaling pathways include the Raf–MEK–Erk kinase cascade, the phosphatidylinositol 3-kinase (PI3K)–Akt pathway, some protein kinase C isoforms, RalGDS family members, and AF-6, Nore1, Mekk1 and Rin1 (reviewed in Campbell et al., 1998; Boettner and Van Aelst, 2002).
Insulin controls glucose homeostasis by directing peripheral tissues to transport and metabolize glucose. Insulin production depends on the proper development and maintenance of the endocrine pancreas. Pancreatic β-cells are developed by a series of differentiation and maturation processes and by cell proliferation. The precise molecular mechanisms governing β-cell development are largely unknown, but insights into these events are being discovered by physiological studies of genetically modified animals and isolated islets. Many signals have been implicated in pancreatic β-cell proliferation and function, including insulin-like growth factor-I (IGF-I), prolactin, growth hormone (GH), glucagon-like peptide-1 (GLP1) and others (Nielsen et al., 2001). However, the specific pathways that mediate development of a normal β-cell mass are poorly understood. Several molecules that are upstream and downstream modulators of Ras have been implicated in β-cell proliferation. Both insulin receptor substrate-2 (IRS-2) (Withers et al., 1998) and cyclin-dependent kinase 4 (Cdk4) (Rane et al., 1999) play critical roles in β-cell physiology, since deletion of either of these intracellular signaling molecules produces severe diabetes in mice. The IGF-I signaling pathway is an important contributor to β-cell mass, since transgenic animals overexpressing IGF-I specifically in β-cells develop islet hyperplasia (George et al., 2002). Conversely, heterozygous animals (+/–) for the IGF-I receptor show a reduction in β-cell mass, and this effect is diminished even more potently by the lack of IRS-2 (Withers et al., 1999). In addition, activation of IRS-2-mediated signaling pathways triggered by IGF-I, but not transfrorming growth factor-α (TGF-α) or epidermal growth factor (EGF), augments pancreatic β-cell proliferation in vitro (Lingohr et al., 2002), further demonstrating a link between IGF-I and IRS-2. More recently, transgenic animals overexpressing the PI3K effector Akt/PKB in β-cells revealed its important contribution to the mitogenic signal (Tuttle et al., 2001). Also, knockout mice for S6K1 (p70s6k), another kinase downstream of PI3K, exhibit a reduction in pancreatic β-cell mass (Pende et al., 2000). Interestingly, transgenic mice expressing v-ras specifically in pancreatic islets develop β-cell apoptosis and diabetes (Efrat et al., 1990). Similarly, c-Myc activation in β-cells induces apoptosis and β-cell mass reduction, whereas suppression of the apoptotic effects of c-Myc reveals a potent proliferative activity that results in neoplasia (Pelengaris et al., 2002). Thus, these various observations demonstrate that a balance among the different signaling pathways activated by Ras is essential for normal β-cell development and function.
Ras cycling between an inactive GDP-bound conformation and an active GTP-bound conformation is controlled by guanine nucleotide-releasing factors (GRFs) and GTPase-activating proteins (GAPs), respectively. Thus, GRFs play a critical role in the cell by allowing signals from the extracellular environment to reach the internal response machinery. Different GRF homologs have been cloned and characterized (Bowtell et al., 1992; Martegani et al., 1992; Fam et al., 1997; Ebinu et al., 1998) and their distinct roles are determined by distinct mechanisms of activation and cell-specific expression. Hence, Sos1 and Sos2 are activated preferentially by binding to docking proteins containing SH2 and SH3 domains, as well as the interaction with the PI3K products. On the other hand, GRF1 and GRF2 can be activated by binding to βγ-subunits of heterotrimeric G proteins coupled to receptors (Mattingly and Macara, 1996) as well as by calmodulin-mediated Ca2+ influxes (Farnsworth et al., 1995; Cullen and Lockyer, 2002). Whereas Sos1 and Sos2 are ubiquitously expressed, expression of GRF1 and GRF2 is limited to brain and a few other tissues (Guerrero et al., 1996). Interestingly, GRF1 and GRF2 exhibit different patterns of expression within the brain (Fernandez-Medarde et al., 2002), suggesting different physiological roles despite the high homology between these factors.
GRF1 is an imprinted gene which is expressed only after birth (Ferrari et al., 1994; Itier et al., 1998). Previous reports have demonstrated that GRF1-deficient mice exhibit defects in learning and memory, although some controversy exists regarding the molecular nature of these impairments (Brambilla et al., 1997; Giese et al., 2001; Tonini et al., 2001). Similar to two other GRF1 knockout strains (Itier et al., 1998; Giese et al., 2001), our mice display a reduced body size and low IGF-I levels. Surprisingly, we have observed that GRF1 knockout mice have low levels of circulating insulin and are glucose intolerant due to a reduction of β-cell mass. Here we report that β-cells express GRF1, and deletion of this Ras-GTP exchange factor causes an impairment of β-cell proliferation and neogenesis. Glucose sensing and insulin production are not affected by the absence of Ras-GRF1 signaling since β-cells from knockout animals secrete and synthesize insulin comparably with control mice. One explanation for the impaired β-cell proliferation observed in GRF1-deficient mice is the failure to activate PI3K–Akt and Erk signaling pathways within these cells. Thus, our data reveal an important role for Ras-GRF1-mediated signaling in the regulation of β-cell proliferation and glucose homeostasis.
Results
To study the physiological role of GRF1, we used gene targeting technology in embryonic stem (ES) cells to generate mutant mice deficient for GRF1 (Figure 1A). A deletion in two exons coding for the catalytic domain inactivated all of the potential splicing isoforms of GRF1 (Cen et al., 1992). As expected, our mutant mice did not express GRF1 (Figure 1B and C) based on Southern analysis and RT–PCR. These results were confirmed further by immunoblotting with GRF1-specific antibodies (Figure 1C). GRF1 knockout animals characterized by others have been reported to display alterations in learning and long-term memory (Brambilla et al., 1997; Giese et al., 2001; Tonini et al., 2001) and we did not study such phenotypes in our animals. On the other hand, in agreement with a previous report (Itier et al., 1998), our GRF1 knockout mice displayed a marked reduction in body size and weight (∼70% of wild-type counterparts; Fernandez-Medarde et al., 2002) without any other observable somatic phenotype. GRF1 is expressed preferentially, but not exclusively, in brain, suggesting its importance in the central nervous system (CNS). However, we have also detected the expression of GRF1 in wild-type testis and faintly in whole pancreas (Figure 1C) as well as in other tissues (Guerrero et al., 1996).

Fig. 1. Generation and verification of the targeted deletion of the mouse GRF1 gene. (A) Protein structure of GRF1 and genomic targeting strategy. Relevant structures including two pleckstrin homology (PH) domains, the DH domain and CDC25-like domain are indicated in the schematic of p140 GRF1. The seven exons encoding amino acid sequences through the CDC25-like catalytic domain were mapped and sequenced in the murine GRF1 gene (vertical bars, arbitrarily assigned I–VII). A 1.8 kb region of GRF1 containing two exons of the catalytic region was replaced by the PGK promoter-driven neomycin cassette. Restriction sites: B, BamHI; H, HindIII; S′, SacI; Sp, SpeI; Sw, SwaI. (B) This modification produced a diagnostic reduction of 6 kb in a SpeI fragment which was detected by Southern blot analysis. (C) Tissue distribution of GRF1 expression in wild-type and knockout mice. RNA from the indicated tissues was isolated and used in an RT–PCR with specific primers from two consecutive exons (V and VI) for the catalytic region of GRF1 gene: 5′-CTGTGTCCCTTACCTGGGGATGTATCTCAC-3′ and 5′-GGCTGGGGGTCGATTTTGTAGGTAGTCTGC-3′. Primers for the housekeeping gene G3PDH were 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. Tissues were also collected for western blot analysis using immunopurified anti-GRF1 antibodies.
Surprisingly, a detailed hormonal study of GRF1–/– mice revealed low serum insulin levels, without significant alterations of glucagon levels (Figure 2A). Other hormones, including prolactin, thyroid-stimulating hormone, testosterone, estradiol and corticosterone did not change. In both male and female GRF1 knockouts, insulin levels are diminished by ∼30%. This reduction in circulating insulin was detected as early as 6 weeks of age and persisted throughout adulthood. Based on this observation, we examined whether GRF1 is expressed in pancreatic β-cells. RT–PCR analysis revealed the presence of full-length GRF1 as well as Sos1 and Sos2 in isolated murine islets (Figure 2B). However, expression of GRF2 was not detected in islets. Furthermore, GRF2 knockout mice have normal insulin levels (unpublished results). Thus, despite the similarities in the structure and mechanisms of activation shared between GRF1 and GRF2, these results indicate that GRF2 is not involved in β-cell development and function. Our findings suggest a potential direct and specific role for GRF1 in β-cell function (see below). Therefore, we focused on elucidating the role of Ras exchange factor in islet physiology.

Fig. 2. Deletion of GRF1 results in low circulating levels of insulin. (A) Analysis of serum insulin and glucagon levels from adult mice under fed conditions. Data presented correspond to the average ± SEM of seven wild-type and seven GRF1–/– animals. Statistical analysis: *P = 0.05; **P = 0.3. (B) Expression of different GEFs as detected by RT–PCR analysis of isolated wild-type islets, liver, brain and gonadal fat.
To assess whether low insulin circulating levels altered glucose metabolism, we first analyzed glucose tolerance in GRF1–/– animals. GRF1-deficient mice displayed a notable intolerance to glucose (Figure 3A), although by the end point of the test (120 min), glucose concentrations were restored to the normal levels of control animals. We also evaluated glucose-stimulated insulin release under similar in vivo conditions. Interestingly, GRF1 knockouts displayed a dampened release of insulin at the early time point (15 min), but circulating insulin levels were comparable with control values at 120 min. These results parallel the behavior of the GRF1 knockouts during the glucose tolerance test, providing an explanation for both the hyperglycemia and the return to normal glucose levels at the end of the test (Figure 3B). Thus, these assays indicate that GRF1-deficient mice are glucose intolerant due to diminished circulating insulin.

Fig. 3. Analysis of glucose tolerance and insulin release. (A) For evaluation of glucose tolerance, male animals (12 weeks of age) were fasted for 14–16 h. Following intraperitoneal injection of glucose (2 mg/g), serum glucose levels were measured by tail bleed at the indicated time points. Data represent the average ± SEM of six wild-type and six GRF1–/– animals. The experiment shown is representative of at least four independent glucose tolerance tests. (B) Glucose-stimulated insulin release was analyzed in vivo using fasted male animals. Animals were injected intraperitoneally with glucose (2 mg/g body weigth) and serum samples were collected by tail bleed at the indicated times. Insulin levels were determined by ELISA. Results are the average ± SEM of four wild-type and four GRF1–/– animals. *P = 0.04; **P = 0.08; ***P = 0.3.
To determine whether the observed insulin deficiency was accompanied by insulin resistance in peripheral tissues, we performed a set of experiments to test insulin sensitivity in fat, liver and skeletal muscle. First, we evaluated the response of GRF1–/– mice during an insulin tolerance test (ITT). GRF1-deficient animals displayed the same sensitivity to insulin as controls; the reduction of blood glucose levels in response to the insulin injection paralleled that of wild-type animals (Figure 4A). Thus, based on this classic test, peripheral tissues of GRF1 knockouts are not resistant to the physiological effects of insulin and, therefore, insulin resistance is produced by a failure in the insulin signaling pathways (Pessin and Saltiel, 2000). The PI3K–Akt signaling pathway is activated by insulin in a broad variety of tissues and participates in the regulation of glucose homeostasis. To evaluate insulin action in GRF1 knockouts at the molecular level, we analyzed insulin signaling in skeletal muscle and liver. Fasted animals were stimulated with a bolus of insulin and tissues were lysed and studied for activation of insulin signaling pathways. Expression and activation of the insulin receptor in GRF1–/– skeletal muscle and liver (Figure 4B) were comparable with levels detected in control animals. Insulin produced a potent activation of Akt/PKB in muscle and liver in wild-type and knockout mice; Erk was potently activated in muscle, but weakly or not activated in liver from both wild-type and knockout mice. However, no differences in this activation were noted between knockouts and controls (Figure 4B). Therefore, the results of these signaling experiments further confirm that peripheral tissues of GRF1-deficient mice respond properly to insulin; these animals simply have less insulin, but this reduction is not accompanied by insulin resistance.

Fig. 4. Assessment of insulin responsiveness. (A) An insulin tolerance test was performed on fed animals at 8–12 weeks of age. Insulin (1 U/kg) was injected intraperitoneally and glucose levels were sampled at the indicated time. The graph represents the average ± SEM of six wild-type and six GRF1–/– animals. Similar results were obtained in two additional insulin tolerance tests. (B) Insulin-stimulated signal transduction was analyzed in tissues from wild-type and knockout animals. Animals were fasted overnight and then treated with a bolus insulin injection (5 U). Non-stimulated controls were subjected to a saline injection. Tissues were lysed and evaluated by western analysis as shown.
During the study of peripheral insulin signaling, we observed that GRF1 knockouts appeared to have less abdominal fat than control animals. Indeed, a careful analysis revealed that the weight of gonadal fat in GRF1–/– mice is reduced ∼30% compared with wild-type males (Figure 5A). The reduction in fat accumulation was not due to a dysregulation of appetite, as food intake was not reduced in these mice (Figure 5B). Adipose tissue modulates metabolic events by secreting bioactive substances that regulate appetite and insulin resistance. Hence, we measured the levels of leptin and resistin, two hormones produced by fat that have been implicated in these processes (Steppan et al., 2001; Friedman, 2002). However, the expression levels of these hormones were similar in the fat of wild-type and GRF1–/– animals (Figure 5C and D).

Fig. 5. Analysis of adipose storage and metabolism. (A) Gonadal fat pads were collected from male animals at 12–16 weeks of age. Weight of fat (wild type, 1.50 ± 0.07 g; knockout, 0.78 ± 0.16 g) was compared with overall body weight (wild type, 40.2 ± 0.7 g; knockout, 29.5 ± 1.5 g) and used to calculate the given ratios. Data represent the average fat:body comparisons ± SEM of four wild-type and six GRF1–/– mice. *P = 0.04. (B) Food intake was monitored daily in groups of wild-type and knockout animals during a period of 4 weeks. Prior to onset of the study, animals were given 1 week to adjust to feeding jars and ground chow. Results represent the average chow consumed per animal per day ± SEM from five wild-type and eight knockout animals. (C) Serum leptin was assessed from fed animals by ELISA. Results represent the average ± SEM circulating leptin levels of five wild-type and eight knockout animals. (D) For analysis of resistin expression, adipose tissue was collected from fasted animals and used for preparation of RNA. RT–PCR was performed to detect resistin expression, and densitometric analysis was used to assess relative levels of resistin from ethidium bromide-stained gels. RNA was prepared and evaluated independently from the fat pads of five wild-type and eight knockout animals. (E) Lipolysis was analyzed in isolated adipocytes of fed male mice (12 weeks of age). The rate of basal, insulin-stimulated (10–8 M), and isoproterenol-stimulated (ISO; 10–6 M) glycerol formation was measured. Data represent the average ± SEM rates of lipolysis of three wild-type and three knockout animals.
One classic action of insulin is to suppress lipolysis in adipose tissue. Thus, given that serum insulin levels are reduced in GRF1–/– animals, we postulated that the reduced fat storage probably reflects failed inhibition of lipolysis in this animal model. Isolated abdominal fat preparations from wild-type and knockout animals were used to determine the lipolytic activity in response to insulin. Interestingly, basal lipolysis levels were almost doubled in GRF1-deficient mice (Figure 5E). However, both groups of animals responded equally well to insulin (Figure 5E) in a dose-dependent manner (data not shown) by blocking lipolysis activity. Additionally, the β-adrenergic agonist isoproterenol induced the lipolytic activity of fat proportionally from both wild-type and GRF1-deficient animals. Thus, the results of the lipolysis study confirm (i) that basal lipolytic activity is higher in adipose tissue of GRF1 knockouts, possibly owing, at least in part, to lower circulating insulin levels; and (ii) that based on the normal response of GRF1–/– fat to insulin, this tissue displays no insulin resistance. Finally, no significant elevations in serum-free fatty acids were detected in GRF1 knockouts (3–5 months of age: 0.9 ± 0.1 mEq/l, wild type; and 0.8 ± 0.2 mEq/l, GRF1–/–), suggesting that free fatty acids liberated by the increased lipolysis in these animals serve as a source of fuel. Taken together, these data indicate that GRF1 knockout mice display abnormalities in fat storage due to enhanced basal lipolysis but not due to differences in food consumption.
The results thus far indicate that GRF1 knockout mice are glucose intolerant due to insulin deficiency. Hence we focused on β-cell physiology to elucidate the nature of the defects underlying the reduction in serum insulin. We reasoned that three different possibilities might account for the hypoinsulinemia observed in the GRF1 knockouts: a reduction in insulin production; a defect in glucose sensing or insulin secretion; and/or an overall reduction in the mass of β-cells. Strikingly, we observed a reduction in β-cell area and islet size (Figure 6A). Based on insulin immunostaining of pancreatic sections taken from animals at 3 months of age, quantification of the β-cell area (percentage of the total pancreas) revealed a 30% reduction in GRF1–/– mice as compared with controls (Figure 6B). Moreover, the average size of GRF1–/– islets was noticeably smaller (Figure 6C). The reduction in size and overall area occupied by β-cells might reflect fewer and/or smaller cells. To distinguish between these possibilities, we analyzed β-cell density within a constant area of islets. 4′,6-diamidino-2-phenylindole (DAPI)-staining nuclei revealed that the number of insulin-positive cells per area was equivalent between GRF1 knockouts and wild-type controls (Figure 6D), demonstrating that deletion of this Ras exchange factor does not alter β-cell size but rather reduces the overall number of cells. As described above, isolated islets from wild-type pancreas contain high levels of GRF1 mRNA, as well as Sos1 and low levels of Sos2. However, expression of GRF2 is not detected in normal islets, and GRF2–/– mice have normal insulin levels (Figure 2B). These observations suggest specifically that the lack of Ras-GRF1 activity may account for the reduction in β-cell area, which underlies the hypoinsulinemia and glucose intolerance in the knockout animals.

Fig. 6. Morphological analysis of pancreatic β-cells. (A) Representative insulin immunostaining of pancreatic sections from wild-type and knockout male mice. Upper panels: low magnification immunohistochemical analysis with anti-insulin antibodies. Lower panels: higher magnification of immunofluorescence studies. (B) The percentage of total pancreatic area occupied by β-cells was calculated using insulin-stained pancreas sections. For this analysis, three sections from each animal were immunostained and data were collected from eight independent fields of each section using Openlabs software. Results represent examination of sections from four wild-type and four knockout animals (16 weeks of age; P = 0.06). (C) Average islet size was assessed using pancreatic sections double-stained with glucagon and insulin. Measurements were performed using Openlabs software. Results represent examination of sections from four wild-type and four knockout animals (16 weeks of age; P = 0.03). (D) To approximate the density of β-cells/islet, paraffin-embedded pancreas sections were immunostained with insulin. To reveal the nuclei, sections were also exposed to DAPI. The area represented by insulin-containing cells was tabulated as described above, and nuclei were counted manually from these cells stained for insulin.
To investigate further the mechanisms by which Ras-GRF1 contributes to β-cell function, we considered the various developmental processes that ultimately determine β-cell number: neogenesis, growth and apoptosis. In mice, formation of the pancreatic bud occurs approximately at embryonic day 9.5 and although islets do not form properly until the end of gestation, insulin-producing cells can be detected in the developing pancreas as early as E10 (Sander and German, 1997). However, GRF1 is not expressed until after birth (Ferrari et al., 1994; Itier et al., 1998), excluding the possibility that this molecule exerts its role during embryonic development of the endocrine pancreas. Thus, we focused our study on postnatal development of β-cell number. The rodent pancreas undergoes major remodeling during the postnatal phase, and apoptosis is a critical mechanism in this process. In the rat, a major wave of apoptosis occurs in the neonatal pancreas between days 2 and 18, but is followed by increased apoptosis just prior to weaning (Scaglia et al., 1997). A positive outcome, i.e. development of a normal β-cell mass, requires a fine balance between growth and apoptosis; a dysregulation of remodeling events could have important effects on β-cell mass. As GRF1 is not fully expressed until after postnatal day 5, we performed our analysis of proliferation and apoptosis at weaning (postnatal day 21).
Apoptotic events within β-cells were investigated at postnatal day 21 by TUNEL assay and activated caspase-3 on pancreatic histological sections, but no differences in rates of apoptosis were observed between knockouts and controls (data not shown). However, quantification of bromodeoxyuridine (BrdU) incorporation from these same samples revealed a significant reduction in the proliferation of GRF1–/– β-cells (Figure 7A), suggesting that Ras-GRF1 functions in the β-cells by mediating proliferative signals. Moreover, fewer neogenic islets were noted in GRF1–/– pancreas sections (Figure 7B). Substantial evidence indicates that β-cell proliferation and/or survival is dependent on IGF-I (George et al., 2002). Thus, we reasoned that IGF-I signaling might be altered in GRF1–/– β-cells. To test this possibility, we stimulated isolated islets from both knockouts and controls with IGF-I and analyzed downstream signaling pathways that can be activated by Ras-GRF1. IGF-I stimulation of GRF1–/– islets resulted in very weak, if any, activation of Akt, in contrast to islets from controls where a robust phosphorylation of this kinase was detected (Figure 7C). Activation of Erk was also practically absent in GRF1–/– isolated islets (Figure 7C). Immunoblotting of lysates with anti-phosphotyrosine antibodies (PY) revealed specific phosphorylation of a band with the expected mobility of the IGF-I receptor upon IGF-I stimulation in both control and GRF1–/– islets (not shown), indicating that the main effect associated with the absence of Ras-GRF1 is a significant impairment of downstream signaling. Interestingly, these data contrast with our earlier observations in peripheral tissues where Akt was activated normally by insulin in skeletal muscle of GRF1–/– mice (Figure 4B). Moreover, under our experimental conditions, no significant differences in apoptosis were detected between wild-type and knockout isolated islets as determined by poly(ADP-ribose) polymerase (PARP) degradation (Tewari et al., 1995) (Figure 7D). However, we have not assessed whether longer periods of serum deprivation might reveal differences between control and knockout islets. These findings suggest that Ras-GRF1 might be required for proper activation of Akt and Erk by IGF-I specifically in the β-cell context. We have not determined sensing of other mitogens that could exert their action independently of GRF1. Recent studies with transgenic animals overexpressing the PI3K effector Akt/PKB in β-cells have revealed that these animals display an increased β-cell mass together with an increased β-cell size (Tuttle et al., 2001). Thus, taken together, our results provide a mechanism by which Ras-GRF1 signaling contributes to the proliferative action of IGF-I in pancreatic β-cells.
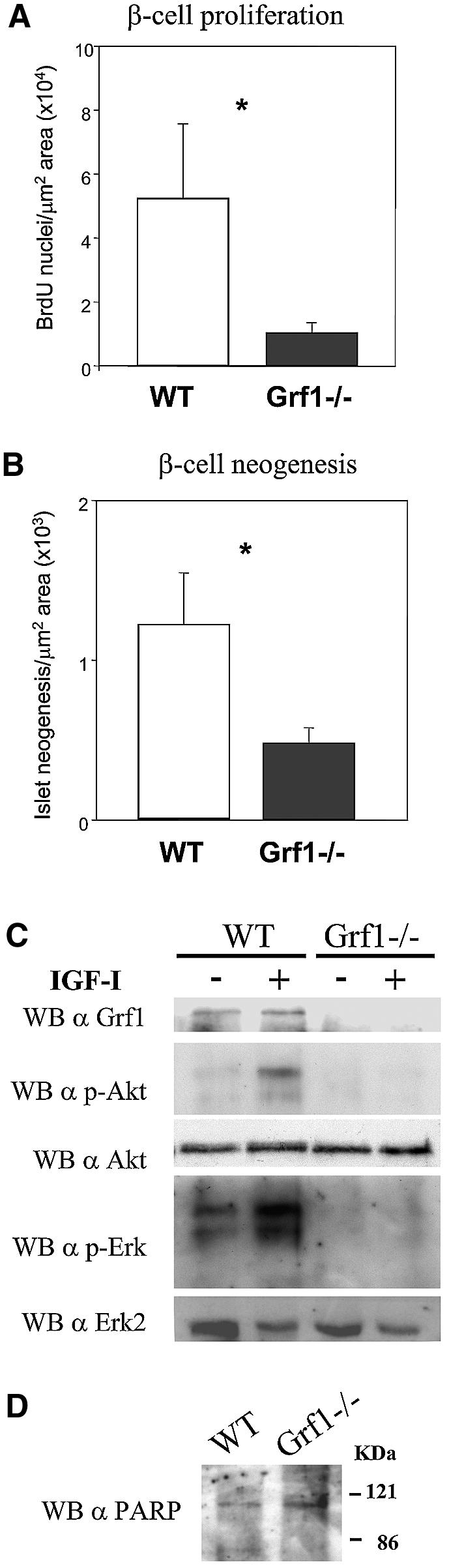
Fig. 7. Evaluation of β-cell growth and IGF-mediated signals. (A) BrdU incorporation was monitored using immunohistochemistry of pancreatic sections prepared from control and knockout mice of 21 days of age. Sections were double-stained with anti-BrdU and anti-insulin. Cells stained for both were scored positive. Three sections from each animal were studied. Data represent the average values ± SEM of three wild-type and three knockout animals. *P = 0.02. (B) Using the same sections as for the study of BrdU incorporation, neogenic islets were tabulated. Islets containing no more than three cells stained positive for insulin were considered neogenic. Data represent the average values ± SEM obtained from three wild-type and three knockout animals. *P = 0.04. (C) Isolated islets were stimulated with 10 nM IGF-I for 15 min in the presence of glucose (RPMI 1640 serum-free medium) and subsequently lysed. Equal amounts of protein were analyzed for activation of MAPK and Akt by western blotting. (D) Isolated islets were tested for apoptosis by means of immunoblotting with anti-PARP antibodies.
Finally, to determine whether the reduction in β-cell area might also be accompanied by functional defects in insulin production and/or release, we analyzed secretion in isolated islets. Islets were carefully size matched and then stimulated in vitro with glucose or arginine. l-arginine stimulates insulin secretion through membrane depolarization, whereas protein kinase A and protein kinase C are implicated in the stimulatory effect of membrane depolarization in glucose-induced insulin secretion (Thams and Capito, 1999). Total insulin content in size- and number-matched isolated islets was equivalent between wild-type and knockout animals (Figure 8A), demonstrating that deletion of GRF1 does not directly impair insulin production. Moreover, insulin secretion in response to various concentrations of glucose and/or arginine was similar in wild-type and knockout islets (Figure 8B). We also tested the β-adrenergic agonist adrenaline to determine the ratio of glucose counteraction between both strains. As indicated in Figure 8B, adrenaline affected both wild-type and knockout islets equally well. These findings indicate that Ras-GRF1 signaling does not exert a primary function in insulin production and secretion but rather this pathway can positively regulate the pool of β-cells. Therefore, reduction in the pancreatic β-cell area observed in GRF1-deficient animals is responsible for the decreased serum insulin levels in these mice. Interestingly, our characterization of the β-cell phenotype of the GRF1 knockouts reveals that they are quite similar to S6K1-deficient mice (Pende et al., 2000). S6K1-null mice have reduced body size, insulin insufficiency and glucose intolerance due to a reduction of β-cell size and mass.

Fig. 8. Functional analysis of pancreatic β-cells. (A) Total insulin content was determined from isolated islets by radioimmunoassay and plotted as µunits of total insulin content per islet. For the experiment, islet pools from the two animal groups were matched carefully by size. Data represent the average ± SEM of three wild-type and three knockout animals (12 weeks of age). (B) Glucose-stimulated insulin release was analyzed using the indicated concentrations of glucose, arginine (10 mM) and adrenaline (1 µM). Insulin concentrations were determined by radioimmunoassay and plotted as µunits of secreted insulin per ml and per islet in 1 h. Data represent the average insulin levels ± SEM of three wild-type and three knockout animals.
Discussion
The endocrine pancreas is subject to dynamic changes in response to variations in the demand for insulin. The β-cell population is kept in balance by β-cell formation (neogenesis and proliferation) and β-cell death. Many of the factors that control this delicate balance have been described. However, the signaling pathways that govern these processes are poorly understood. Our results provide a new physiological role for Ras-GRF1 signaling in β-cell proliferation and neogenesis. GRF1 is an imprinted gene regulated by promoter DNA methylation (Yoon et al., 2002) that is expressed after birth (Ferrari et al., 1994; Itier et al., 1998). Although GRF1 is expressed preferentially in the CNS, we show that pancreatic islets also express GRF1, but not GRF2, suggesting a direct role for Ras-GRF1 physiology in islet function. Indeed, our GRF1-deficient mice display a 30% reduction in β-cell area due to impaired β-cell proliferation and neogenesis. Islet insulin content and insulin secretion were unaltered by the absence of Ras-GRF1 signaling. Thus, GRF1 specifically participates in proliferative signals required for development or maintenance of β-cells. GRF1-null mice are hypoinsulinemic and glucose intolerant, but not insulin resistant, as predicted from ITT experiments and normal activation of insulin signaling pathways in peripheral tissues.
Regulation of glucose homeostasis by insulin depends on the maintenance of β-cell mass and function. GRF1-deficient mice display abnormalities in glucose metabolism. The low circulating insulin levels correlate with an increased lipid catabolism in fat, which is rapidly disposed of or used as fuel, since no variation in free fatty acids was observed in serum when compared with wild-type animals. Alternatively, it is possible that the absence of Ras-GRF1 expression in brain and/or in fat results in alterations in brain-released circulating hormones or sensing of them in the fat, resulting in the overall increased lipolysis found in GRF1 knockouts. Interestingly, despite the reduced insulin levels and glucose intolerance, GRF1 knockouts do not develop diabetes. We have studied glucose homeostasis in these animals up to 1 year of age and have observed no progression to frank diabetes (data not shown). Ultimately, the low fat content, the lack of peripheral insulin resistance and the reduced body size are probably important factors that allow GRF1–/– animals to compensate for insulin deficiency and prevent the development of diabetes.
In GRF1 knockout mice, no acceleration of β-cell apoptosis was detected, whereas the rate of β-cell proliferation is markedly reduced. We also observed lower β-cell neogenesis during the phase of major islet organization at 21 days after birth. GRF1 knockout animals have a reduced body size but they do not display a reduction in β-cell size. Interestingly, S6K1-deficient animals are also smaller, with a β-cell mass reduced to an extent similar to the GRF1-deficient mice. However, reduction in β-cell mass in S6K1 knockout mice is due to a reduced β-cell size, in contrast to our GRF1 knockout mice. We show that in the absence of GRF1, IGF-I stimulation does not activate Akt in islets. Overexpression of activated Akt1 in β-cells results in an increased β-cell mass together with an increased β-cell size (Tuttle et al., 2001). Ras effectors include PI3K and others. The Raf–MEK–Erk signaling pathway is also activated by Ras. As previously suggested (Lingohr et al., 2002), it is possible that the equilibrium of activation of the different signaling pathways emanating from Ras, such as Erk and Akt, may result in different responses and functions.
Ras physiology and development have been broadly studied in different biological models. However, in knockout mice, H-Ras and N-Ras, two of the three Ras isoforms, are dispensable for normal growth and development (Esteban et al., 2001). In contrast, K-Ras has an essential role during development. Furthermore, the majority of Ras mutations described in tumors are found in K-Ras (Ellis and Clark, 2000), suggesting its preferential role in cell growth control. Interestingly, Sos1 is the only Ras-GRF essential for life (Wang et al., 1997; Qian et al., 2000). It is possible that these redundant forms of Ras and their activating genes may reflect a step in evolution in which more defined roles have evolved for each of the genes. On the other hand, the different mechanisms of activation of the GRF subfamilies may reflect their distinctive physiological roles.
Our results provide a novel physiological role for GRF1 in β-cell development; based on the characterization of insulin deficiency in GRF1 knockouts, this Ras exchange factor is essential for postnatal proliferation of β-cells. The homeostatic control of β-cell mass in both normal and pathophysiological conditions is based on the balance of cell proliferation, cell growth and cell death. A dysregulation of this balance would be expected to have negative consequences for β-cell mass and, thus, for glucose homeostasis. Diabetes (both type 1 and type 2) is characterized by insulin insufficiency. A full understanding of the molecular mechanisms that regulate β-cell proliferation and death are required for the development and implication of new strategies to treat and prevent β-cell failure. Thus, the GRF1 knockout model now provides an opportunity to determine how Ras-GRF1 pathways may modulate mitogenic signaling in pancreatic β-cells.
Materials and methods
Generation of GRF1 knockouts
λ genomic clones containing several exons of the CDC25-H domain of GRF1 were isolated from a 129 SVJ mouse genomic library (Stratagene). Our GRF1 gene targeting vector was constructed by substituting a SwaI–BamHI 1.8 kb genomic fragment for a neomycin cassette driven by the phosphoglycerate kinase 1 promoter (PGKneo), which was employed as a positive selectable marker. The PGK–thymidine kinase cassette was used as a negative selectable marker (Tessarollo, 2001) in our vector. Electroporation of the RW4 embryonic stem (ES) cell line and selection of transfected clones was performed as described elsewhere (Tessarollo, 2001). DNAs derived from G418/FIAU-resistant ES clones were screened by PCR and Southern blotting by means of SpeI restriction enzyme digestion with a 5′ probe external to the targeting vector sequence, as indicated in Figure 1A. Recombinant clones containing the predicted 6.6 kb rearranged band were obtained at a frequency of 1/33. Independent, targeted ES cell clones for the GRF1 gene injected into C57BL/6N blastocysts generated chimeras that transmitted the mutated allele to the progeny (Bonin et al., 2001). Mice were bred in a specific pathogen-free facility, with food and water provided ad libitum. Breeding heterozygous animals obtained from chimera crossings gave rise to homozygous mutant GRF1–/– mice at normal Mendelian proportions. Proper insertion of the targeting construct was monitored by Southern blotting with SpeI-digested DNA from tail biopsies using a SpeI–HindIII probe 5′ from exon IV (see Figure 1B). Genotyping of mice or ES cells was performed by PCR with the primers 5′-AGTTGCTGAAGGGAGG AGGGTAGCGGC-3′, 5′-TGACCAGGCCGTCCTCTGTGTAATTGG-3′ and 5′-CTACCGGTGGATGTGGAATGTGTGCGA-3′ (the latest hybridizing to the neo-pgk promoter), resulting in a 824 bp band for the wild type and a 615 bp band for the knockout.
RT–PCR analysis
Isolation of RNA was performed by homogenization of tissues in TRIZOL (Gibco-BRL) following the manufacturer’s indications. Isolated RNA was submitted to DNase digestion followed by a second purification through Qiagen columns (RNeasy kit). A 0.5 µg aliquot of total RNA was used in an RT–PCR (Titan One Tube kit; Roche) using the following primers: 5′-AACATACACTCACCCATATCTCCCTTCGGC-3′ and 5′-TGCGGTTATTGTCCACCAGATCACGACACG-3′ for GRF1; 5′-GTGAGGGCCAGAAAGCTGTCTTTGACGTCT-3′ and 5′-TCGGCT ACCTGTCCTCCAGGCCTGCCGATT-3′ for GRF2; 5′-AGAAGC TGATCGCATAGCTATAGAGAATGG-3′ and 5′-CAGGTCTGCTTA TGTGCCACTCAACTGTGG-3′ for Sos1; 5′-GAACCTACTGAGG CAGACAAGCTGGCGTTA-3′ and 5′-CGAACTGTCCTGTTCTACT GATGTGCCAT-3′ for Sos2; and 5′-TCATTTCCCCTCCTTTTCCT-3′ and 5′-CAAGACTGCTGTGCCTTCTG-3′ for resistin.
Isolation of islets
Pancreas were collected from sacrificed animals, carefully minced in Hank’s balance salt solution (HBSS) buffer and digested with collagenase P (Boehringer Mannheim) for 15 min. Digested tissue was washed once with HBSS and resuspended in 25% Ficoll in a 15 ml plastic tube. On top of this suspension, a discontinuous density gradient of 23% (3 ml), 20% (2 ml) and 11% (2 ml) Ficoll solutions was loaded. After 20 min of centrifugation at 1000 g, the interface between 11 and 20% Ficoll was collected and washed twice with HBSS. Finally, islets were resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Cultured islets were left to recover for 1 day and then transferred to RPMI 1640 supplemented with 0.5% FBS. Stimulation with IGF-I was performed next morning in RPMI 1640 serum-free medium.
Metabolic studies
Animals were maintained on a normal light/dark cycle and handled in accordance with European Community protocols for animal studies. Serum glucose levels were measured from mouse tails using a Glucometer Elite glucometer (Bayer). We obtained blood for plasma insulin levels either by tail bleeds or upon sacrifice of animals. Immunoreactive insulin levels were measured either by radioimmunassay using rat insulin (Linco) as a standard or by enzyme-linked immunosorbent assay (ELISA; CrystalChem) with mouse insulin as a standard. Leptin was assayed by ELISA (CrystalChem). We performed glucose tolerance tests on animals after a 14–16 h overnight fast. For these studies, glucose (2 mg of glucose per g of body weight) was injected intraperitoneally. Insulin tolerance was evaluated in fed animals injected with 1 U of insulin/kg.
Immunoprecipitation and western blot analysis
Animals were fasted overnight and, following administration of anesthesia, were injected with a bolus of insulin via the inferior vena cava (5 U). After 10 min incubation, liver and soleus muscle tissue were removed and frozen in liquid nitrogen. Non-stimulated controls received an injection of saline. Tissues subsequently were lysed and homogenates were allowed to solubilize for 1 h at 4°C and clarified by centrifugation at 15 000 r.p.m. for 30 min. For analysis of insulin receptor expression, supernatants containing total protein (2 mg) were immunoprecipitated and subsequently probed with anti-insulin receptor antibody (Santa Cruz Biotech.). For analysis of mitogen-activated protein kinase (MAPK) and Akt activation, equal amounts of tissue lysate (50 µg) were probed with anti-phospho-MAPK (Cell Signaling) or anti-phospho-Akt (specific for phosphoSer473; Cell Signaling). Other antibodies used were polyclonal immunopurified anti-GRF1 antibodies, anti-PARP antibodies (C210; Enzyme System Products) and anti-phosphotyrosine antibodies (PY20; Santa Cruz Biotech.).
Immunohistochemistry of pancreas and quantitation of β-cells
For analysis of adult pancreases, animals were killed by overdose of sodium amytal. Each pancreas was removed, cleared of fat and spleen, weighed and fixed overnight in Bouin’s solution. Tissues were embedded in paraffin, and consecutive sections (5 µm) were mounted on slides. Following re-hydration and permeabilization (0.1% Triton X-100), sections were immunostained for α-cells using mouse monoclonal anti-glucagon antibodies (Sigma). β-cells were either immunostained using guinea pig anti-insulin (Dako Corp.) and processed for immunohistochemistry with Vectastain Elite kit (Vector Laboratories) using 3,3′-diaminobenzidine as chromagen, or alternatively they were stained with mouse anti-insulin antibodies (Sigma) and processed for immunofluorescence staining using fluorescein isothiocyanate (FITC)-coupled anti-mouse antibodies (Jackson Immunochemicals). Detection was performed using rhodamine and fluorescein antibodies (Jackson Immunoresearch). Sections were incubated briefly in DAPI (0.01%) to reveal total cell nuclei. For measuring β-cell area, sections were viewed using a Zeiss Axiovert S100 TV microscope and video camera at a magnification of ×20. Three sections of each pancreas were covered systematically by accumulating images from eight non-overlapping fields. Analyses of cell area and size were performed using Openlab image analysis software (Improvision Imaging). This software calibrated the magnification of each micrograph and tabulated the percentage of pancreatic area positive for insulin reactive cells.
Insulin content of islets and glucose-stimulated insulin release using isolated islets
Islets isolated by collagenase P digestion were hand picked (Jonas et al., 1998) and further pre-incubated at 37°C for 1 h in a control medium containing 15 mM glucose, a concentration that causes half-maximal stimulation of insulin secretion in mouse islets. The islets were then incubated for 60 min, in batches of three, in 1 ml of medium containing the appropriate concentrations of the test substances. At the end of the incubation, a portion of the medium was withdrawn and diluted before insulin assay measurement was performed by double antibody radioimmunoassay (CIS Radioquímica-Schering, Spain).
Lipolysis assay
White adipose tissue of perirenal and epididymal origin was removed from fed mice. Isolated fat cells were obtained by collagenase P digestion (1.5 mg/ml) of adipose tissue fragments in Krebs–Ringer–bicarbonate buffer containing albumin (3.5 g/100 ml) (KRBA) and glucose (6 mM) adjusted to pH 7.4 just before use. After 30 min of digestion at 37°C under shaking, isolated cells were filtered and washed three times in KRBA buffer to eliminate collagenase. The packed cells were adjusted to a suitable dilution in the same buffer. Fat cells were incubated in plastic vials (1 ml of incubation medium) with gentle shaking at 37°C under an air phase. After 90 min incubation, the tubes were placed on ice and 200 µl aliquots were removed for the determination of glycerol, taken as an index of fat cell lipolysis. Metabolic activity is expressed as micromoles of glycerol produced per 100 mg of total lipid, determined gravimetrically after extraction. Pharmacological agents were added just before the incubation was started. When isoproterenol was used, ascorbic acid (0.1 mM) was included in the medium.
Monitoring food consumption
Standard feeding jars containing ground chow were placed in cages of study animals. Prior to onset of measurements, animals were allowed 1 week to adjust to the new system of food acquisition. On day 1 of study, animals were weighed and a given quantity of chow was placed in the feeding jars. Chow consumption was assessed every 2 days by weighing the feeding containers. Animals were weighed weekly. Food intake was monitored for a 4 week period.
Acknowledgments
Acknowledgements
We thank Celso Collazo for technical assistance. This work was supported by FEDER grants 1FD97-1735 and 1FD97-1678 and CICYT grant SAF00-0069 from MCYT (Spain). J.F.d.M. and D.J.B. were financed by the Ramón y Cajal Program from MCYT (Spain).
References
- Boettner B. and Van Aelst,L. (2002) The RASputin effect. Genes Dev., 16, 2033–2038. [DOI] [PubMed] [Google Scholar]
- Bonin A., Reid,S.W. and Tessarollo,L. (2001) Isolation, microinjection and transfer of mouse blastocysts. Methods Mol. Biol., 158, 121–134. [DOI] [PubMed] [Google Scholar]
- Bowtell D., Fu,P., Simon,M. and Senior,P. (1992) Identification of murine homologues of the Drosophila son of sevenless gene: potential activators of ras. Proc. Natl Acad. Sci. USA, 89, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R. et al. (1997) A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature, 390, 281–286. [DOI] [PubMed] [Google Scholar]
- Campbell S.L., Khosravi-Far,R., Rossman,K.L., Clark,G.J. and Der,C.J. (1998) Increasing complexity of Ras signaling. Oncogene, 17, 1395–1413. [DOI] [PubMed] [Google Scholar]
- Cen H., Papageorge,A.G., Zippel,R., Lowy,D.R. and Zhang,K. (1992) Isolation of multiple mouse cDNAs with coding homology to Saccharomyces cerevisiae CDC25: identification of a region related to Bcr, Vav, Dbl and CDC24. EMBO J., 11, 4007–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen P.J. and Lockyer,P.J. (2002) Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell. Biol., 3, 339–348. [DOI] [PubMed] [Google Scholar]
- Ebinu J.O., Bottorff,D.A., Chan,E.Y., Stang,S.L., Dunn,R.J. and Stone,J.C. (1998) RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science, 280, 1082–1086. [DOI] [PubMed] [Google Scholar]
- Efrat S., Fleischer,N. and Hanahan,D. (1990) Diabetes induced in male transgenic mice by expression of human H-ras oncoprotein in pancreatic β-cells. Mol. Cell. Biol., 10, 1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C.A. and Clark,G. (2000) The importance of being K-Ras. Cell Signal., 12, 425–434. [DOI] [PubMed] [Google Scholar]
- Esteban L.M. et al. (2001) Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol., 21, 1444–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fam N.P., Fan,W.T., Wang,Z., Zhang,L.J., Chen,H. and Moran,M.F. (1997) Cloning and characterization of Ras-GRF2, a novel guanine nucleotide exchange factor for Ras. Mol. Cell. Biol., 17, 1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth C.L., Freshney,N.W., Rosen,L.B., Ghosh,A., Greenberg, M.E. and Feig,L.A. (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature, 376, 524–527. [DOI] [PubMed] [Google Scholar]
- Fernandez-Medarde A., Esteban,L.M., Nunez,A., Porteros,A., Tessarollo,L. and Santos,E. (2002) Targeted disruption of Ras-Grf2 shows its dispensability for mouse growth and development. Mol. Cell. Biol., 22, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari C., Zippel,R., Martegani,E., Gnesutta,N., Carrera,V. and Sturani,E. (1994) Expression of two different products of CDC25Mm, a mammalian Ras activator, during development of mouse brain. Exp. Cell Res., 210, 353–357. [DOI] [PubMed] [Google Scholar]
- Friedman J. (2002) Fat in all the wrong places. Nature, 415, 268–269. [DOI] [PubMed] [Google Scholar]
- George M., Ayuso,E., Casellas,A., Costa,C., Devedjian,J.C. and Bosch,F. (2002) β-cell expression of IGF-I leads to recovery from type 1 diabetes. J. Clin. Invest., 109, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K.P., Friedman,E., Telliez,J.B., Fedorov,N.B., Wines,M., Feig,L.A. and Silva,A.J. (2001) Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1). Neuropharmacology, 41, 791–800. [DOI] [PubMed] [Google Scholar]
- Guerrero C., Rojas,J.M., Chedid,M., Esteban,L.M., Zimonjic,D.B., Popescu,N.C., Font de Mora,J. and Santos,E. (1996) Expression of alternative forms of Ras exchange factors GRF and SOS1 in different human tissues and cell lines. Oncogene, 12, 1097–1107. [PubMed] [Google Scholar]
- Itier J.M. et al. (1998) Imprinted gene in postnatal growth role. Nature, 393, 125–126. [DOI] [PubMed] [Google Scholar]
- Jonas J.C., Gilon,P. and Henquin,J.C. (1998) Temporal and quantitative correlations between insulin secretion and stably elevated or oscillatory cytoplasmic Ca2+ in mouse pancreatic β-cells. Diabetes, 47, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Lingohr M.K., Dickson,L.M., McCuaig,J.F., Hugl,S.R., Twardzik,D.R. and Rhodes,C.J. (2002) Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-α or EGF, augments pancreatic β-cell proliferation. Diabetes, 51, 966–976. [DOI] [PubMed] [Google Scholar]
- Martegani E., Vanoni,M., Zippel,R., Coccetti,P., Brambilla,R., Ferrari,C., Sturani,E. and Alberghina,L. (1992) Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. EMBO J., 11, 2151–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattingly R.R. and Macara,I.G. (1996) Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein βγ subunits. Nature, 382, 268–272. [DOI] [PubMed] [Google Scholar]
- Nielsen J.H., Galsgaard,E.D., Moldrup,A., Friedrichsen,B.N., Billestrup,N., Hansen,J.A., Lee,Y.C. and Carlsson,C. (2001) Regulation of β-cell mass by hormones and growth factors. Diabetes, 50, S25–S29. [DOI] [PubMed] [Google Scholar]
- Pelengaris S., Khan,M. and Evan,G.I. (2002) Suppression of Myc-induced apoptosis in β-cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell, 109, 321–334. [DOI] [PubMed] [Google Scholar]
- Pende M., Kozma,S.C., Jaquet,M., Oorschot,V., Burcelin,R., Le Marchand-Brustel,Y., Klumperman,J., Thorens,B. and Thomas,G. (2000) Hypoinsulinaemia, glucose intolerance and diminished β-cell size in S6K1-deficient mice. Nature, 408, 994–997. [DOI] [PubMed] [Google Scholar]
- Pessin J.E. and Saltiel,A.R. (2000) Signaling pathways in insulin action: molecular targets of insulin resistance. J. Clin. Invest., 106, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Esteban,L., Vass,W.C., Upadhyaya,C., Papageorge,A.G., Yienger,K., Ward,J.M., Lowy,D.R. and Santos,E. (2000) The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J., 19, 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane S.G., Dubus,P., Mettus,R.V., Galbreath,E.J., Boden,G., Reddy,E.P. and Barbacid,M. (1999) Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in islet β-cell hyperplasia. Nat. Genet., 22, 44–52. [DOI] [PubMed] [Google Scholar]
- Sander M. and German,M.S. (1997) The β-cell transcription factors and development of the pancreas. J. Mol. Med., 75, 327–340. [DOI] [PubMed] [Google Scholar]
- Scaglia L., Cahill,C.J., Finegood,D.T. and Bonner-Weir,S. (1997) Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology, 138, 1736–1741. [DOI] [PubMed] [Google Scholar]
- Steppan C.M., Bailey,S.T., Bhat,S., Brown,E.J., Banerjee,R.R., Wright,C.M., Patel,H.R., Ahima,R.S. and Lazar,M.A. (2001) The hormone resistin links obesity to diabetes. Nature, 409, 307–312. [DOI] [PubMed] [Google Scholar]
- Tessarollo L. (2001) Manipulating mouse embryonic stem cells. Methods Mol. Biol., 158, 47–63. [DOI] [PubMed] [Google Scholar]
- Tewari M., Quan,L.T., O’Rourke,K., Desnoyers,S., Zeng,Z., Beidler,D.R., Poirier,G.G., Salvesen,G.S. and Dixit,V.M. (1995) Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell, 81, 801–809. [DOI] [PubMed] [Google Scholar]
- Thams P. and Capito,K. (1999) l-arginine stimulation of glucose-induced insulin secretion through membrane depolarization and independent of nitric oxide. Eur. J. Endocrinol., 140, 87–93. [DOI] [PubMed] [Google Scholar]
- Tonini R. et al. (2001) Involvement of CDC25Mm/Ras-GRF1-dependent signaling in the control of neuronal excitability. Mol. Cell. Neurosci., 18, 691–701. [DOI] [PubMed] [Google Scholar]
- Tuttle R.L., Gill,N.S., Pugh,W., Lee,J.P., Koeberlein,B., Furth,E.E., Polonsky,K.S., Naji,A. and Birnbaum,M.J. (2001) Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat. Med., 7, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Wang D.Z., Hammond,V.E., Abud,H.E., Bertoncello,I., McAvoy,J.W. and Bowtell,D.D. (1997) Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for Sos1 in EGFR signaling and development. Genes Dev., 11, 309–320. [DOI] [PubMed] [Google Scholar]
- Withers D.J. et al. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature, 391, 900–904. [DOI] [PubMed] [Google Scholar]
- Withers D.J., Burks,D.J., Towery,H.H., Altamuro,S.L., Flint,C.L. and White,M.F. (1999) Irs-2 coordinates Igf-1 receptor-mediated β-cell development and peripheral insulin signalling. Nat. Genet., 23, 32–40. [DOI] [PubMed] [Google Scholar]
- Yoon B.J., Herman,H., Sikora,A., Smith,L.T., Plass,C. and Soloway,P.D. (2002) Regulation of DNA methylation of Rasgrf1. Nat. Genet., 30, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]