

Abstract
We propose a stepwise mutation model to describe the dynamics of DNA fingerprint variation in Mycobacterium tuberculosis. The genome of M. tuberculosis carries insertion sequences (IS6110) that are relatively stable over time periods of months but have an observable transposition rate over longer time scales. Variability in copy number and genomic location of (IS6110) can be harnessed to generate a DNA fingerprint for each strain, by digesting the genome with a restriction enzyme and using a portion of the element as a probe for Southern blots. The number of bands found for a given genome approximates the number of copies of IS6110 it carries. A large data set of such fingerprints from tuberculosis (TB) cases in San Francisco provides an observed distribution of IS6110 copy number. Implementation of the model through deterministic and stochastic simulation indicates some general features of IS/TB dynamics. By comparing observations with outcomes of the model, we conclude that the IS/TB system is very heterogeneous and far from equilibrium. We find that the transposition parameters have a much stronger effect than the epidemic parameters on copy number distribution.
Efficient molecular methods have allowed the recent genetic characterization of parasitic agents. Such empirical work greatly enhances our understanding of the epidemiology of human diseases (1–4). These new data are the stimulus for the development of theoretical and quantitative understanding of how genetic variability among strains interacts with epidemic processes. The molecular epidemiology of tuberculosis (TB) has been well developed. The insertion sequence (IS) 6110 in the genome of Mycobacterium tuberculosis is stable on the short time scale of months, while transposing at an observable rate over longer time periods. By digesting the genome with the restriction enzyme PvuII and using a portion of the element as a probe for Southern blotting, strains can be distinguished by the resulting DNA fingerprints (2). The number of bands in each blot indicates the number of copies of IS in the isolated genome.
The essential features of TB dynamics recently have been explored (5–7). Those studies take into account the various important aspects of the disease; for instance, a fraction of newly acquired infections progress to the disease immediately while the remainder enter a potentially long period of latent infection. Using estimates of epidemiological parameters from the empirical literature, those authors quantitatively characterize the length of epidemics and assess the efficacy of control strategies.
Several theoretical studies have investigated transposon dynamics. Many of these are constructed for diploid genomes, particularly Drosophila (8–10). Sawyer and Hartl (11) and Moody (12) treat stochastic models of transposable elements in prokaryotes (IS elements), though without consideration of epidemic circumstances. Both studies allow a degree of horizontal transmission. The latter includes replicative transposition (increase by one copy) and deletion (decrease by one copy), whereas the former ignores deletions, arguing that they are much rarer than transposition events. These models bear formal similarities to simple stepwise mutation models for microsatellites in which repeat scores may shift up or down by a single step at a time (13–17).
This study aims to treat the dynamics of IS elements and TB simultaneously. We propose a model that combines the particulars of TB epidemiology with IS transposition as a stepwise process. We discuss to what extent our model can explain the observed distribution of IS copy numbers and draw qualitative conclusions regarding the forces that shape copy number distribution.
The Model
General Description, Assumptions.
This study primarily considers a deterministic model describing the dynamics of IS elements in a TB epidemic. We follow four classes of states: susceptible (S); latently infected but not infectious (L); active infectious cases (T); and recovered individuals (E). Within the L and T classes, we differentiate hosts by the genotype of the strain they carry. Li is the number of individuals latently infected with TB carrying a strain with i copies of the element, and Ti is the number of diseased (infectious) individuals carrying a strain with i copies, where i ≥ 0. These variables are population densities that change over time (t). In our model each host carries at most one strain.
We assume a homogeneous population of constant size, N. All deaths are balanced exactly by births or immigration of susceptible individuals. Let βi be the transmission coefficient for a strain with i copies of the element. The number of new infections associated with an i-copy strain produced per unit time is βiTiS. A fraction p of new cases progresses immediately to the active state, while 1−p of new infections enter the latent state. Let v be the rate at which latently infected individuals progress to the disease per unit time. The copy number of the strain associated with a newly infected susceptible is the same as that of the strain transmitted by the infectious individual in an encounter. We assume there is no superinfection (reinfection) or coinfection. In other words, there is complete cross-protection among hosts infected with different strains.
Mortality is modeled in the following manner: μ is the per capita death rate per unit time for susceptible and latently infected individuals, and μ + μT is the death rate for individuals who have progressed to the disease. Active cases recover at the rate φ per unit time.
We now turn to the transposition-related parameters. A fraction ωi of cases associated with a strain carrying i copies undergoes a change in copy number per unit time. The change is to i − 1 or i + 1 copies. This subsumes the process of within-host substitution. We use the function ωi = 1 − (1 − ω1)i, which combines the events of multiple transpositions in a unit time into a single event. This ensures that ωi never exceeds unity. The parameter ω1 is the rate at which a transposition event takes place in a strain with a single copy. The fractions of events leading to replicative transposition (gain by a copy) and deletion (loss of a copy) are denoted by γ and (1 − γ), respectively. We allow a degree of horizontal transfer of the element h, which is the rate at which strains acquire a copy of the IS element from an external source (biologically, from a different strain). We then can compute the rates of substitution to a copy number higher by one element, bi = γωi + h, and lower by one element, ai = (1 − γ)ωi.
We set a cost, c, to carrying IS elements, reflecting not only the metabolic cost but the increased likelihood of damage to the operation of a cell by disruptive insertion in or near coding regions, or by deleterious chromosomal rearrangements. We subject the transmission parameter to the cost in the following manner: βi = β0(1 − c)i where i is the copy number and 0 < c < 1.
The process we model is represented in Fig. 1. The deterministic dynamics can be represented by the following differential equations.
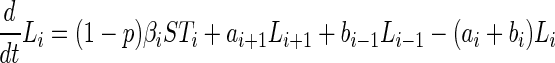 |
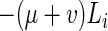 |
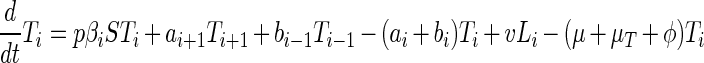 |
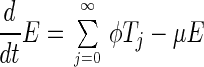 |
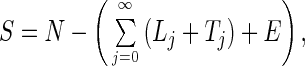 |
1 |
where bi = 0, Li = 0, Ti = 0 for i < 0.
Figure 1.

A scheme for the model. See text for a description of parameters and the dynamical equations (Eq. 1).
Overall Dynamics.
We can find basic epidemic properties of the SLTE system by combining the latent classes (defining X = Σj=0∞Lj) and similarly, the diseased classes (defining Y = Σj=0∞Tj). Define β̄(t) = Σj=0∞βjTj/Y. This quantity is the average transmission coefficient (weighted by the densities of active cases) at a given time. Because βi is a decreasing function of i, 0 ≤ β̄(t) ≤ β0. The following relations describe the overall dynamics of the system:
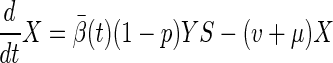 |
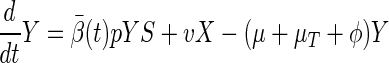 |
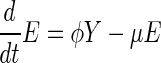 |
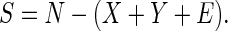 |
2 |
Apart from the fact that β̄(t) is not constant over time, this dynamic is very similar to the model presented in Blower et al. (6, 7) and Porco and Blower (5). The basic reproductive value in this process can be written as a sum of two components corresponding to the “fast” (immediately active) and “slow” (first entering latent stage) pathways of TB infection:
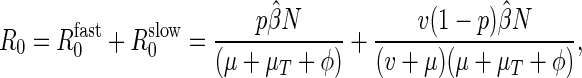 |
3 |
where β̂ is the limit of β̄(t) as t → ∞. It can be shown (see ref. 5) that the exact criterion for the epidemic to be initiated is R0 > 1.
In the absence of mutation, each strain is associated with a basic reproductive value, R0,i, which is identical in form to Eq. 3, with βi = β0(1 −c)i replacing β̂. We can derive a threshold number of elements, iT, below which strains are able to invade a susceptible population. The threshold occurs where R0,i = 1. That is, iT is such that
 |
4 |
Solving for i, we have
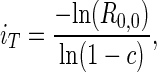 |
5 |
where R0,0 is the basic reproductive value of strains lacking IS elements in the absence of horizontal transfer and transposition. The larger the magnitude of selection against the element (c), the lower the threshold copy number. When c is small, as we presume it to be, iT is very sensitive to changes in its value.
Numerical Investigation: Deterministic Iteration.
Behavior of distribution dynamics.
We implemented a deterministic computer simulation of Eq. 1 by using the Euler method. In this section we choose one set of values for the parameters to demonstrate the appearance of the transient dynamics. We imposed an artificial upper boundary at 400 copies, which is well beyond the observed maximum copy number. The epidemic parameters (v, p, μ, μT, and φ) were chosen from the ranges given in Blower et al. (7). These values are as follows: v = 0.004; p = 0.05; μ = 0.02; μT = 0.1; φ = 0.058. The population size N was set to 500,000; R0,0 was set to be 6, and β0 was chosen to satisfy the relation (Eq. 3), with β0 replacing β̂. We chose to specify R0,0 rather than β0 to ensure that the epidemic is able to establish itself, so that the copy number distribution can be assessed. The copy number of the strain that initiates the epidemic (i0) is 10.
The transposition parameters ω1 and γ can be estimated by using data in Yeh et al. (18) and Niemann et al. (19). In time units of years, these studies yield estimates of ω1 of 0.017 and 0.014 per element per year, respectively. Naas et al. (20) estimate IS-related rates of change in Escherichia coli to be 0.08 per year per culture, which yields ω1 ≃ 0.008, when divided by the typical copy number of 10 (see table 2 in ref. 20). We note that our inferred ω1 in IS6110/TB is probably an overestimate, because some of the altered fingerprint patterns suggest mixed populations (18), and the newly formed strains may not be sufficiently abundant to be transmitted to the next potential host. For simplicity, our model allows instantaneous substitution of strains within hosts rather than allowing mixed populations. Therefore, assuming the estimated values to be upper estimates of ω1, we use ω1 = 0.005. Combining data in Yeh et al. (18) and Niemann et al. (19) gives γ = 0.643, and recognizing again that the issue of mixed populations and the fact that additions are easier to detect than deletions suggest that the actual value is overestimated, we use 0.6.
There are no data for estimating the selection against the IS elements, c, but we believe that it cannot be very high, otherwise variation in the fingerprints would quickly be eliminated. We set it to 0.001. Again, data are unavailable for estimating h, but because gene exchange is known to be very rare in M. tuberculosis (21, 22), we set this to 0.001, making it much lower than ω1.
Numerical iteration of this process using these parameter values reveals the following behavior (see Fig. 2). As the epidemic escalates, the copy numbers diversify, forming a peak near the initial copy number. The peak exists transiently and is eventually lost. The strain with zero copies, that is, the strain lacking IS elements, begins to dominate the population. In the long run, the number of strains carrying IS elements decreases, and IS becomes almost extinct. The equilibrium distribution is highly skewed, with most of the strains lacking IS elements.
Figure 2.

Deterministic numerical iteration. Parameter values used are as follows: i0 = 10; n = 500,000; R0,0 = 6; ν = 0.004; p = 0.05; μ = 0.02; μT = 0.1; φ = 0.058; h = 0.001; ω1 = 0.005; γ = 0.6; c = 0.001. The horizontal axis omits the uninformative high-copy classes (>100). The vertical axis indicates numbers (Ti) of active cases.
Uncertainty and sensitivity analyses.
To assess the effects of the epidemic and transposition parameters on copy number distribution for a range of different parameter values, we make use of the numerical methods of uncertainty and sensitivity analyses (23). Latin hypercube sampling is used to select parameter values. This method divides the specified distribution of each parameter into equiprobable intervals. Each interval is sampled exactly once (without replacement) to form sets of parameters to be used in simulations. This is an efficient method for selecting parameters that reduces the number of simulation runs required to explore the parameter space. Sensitivity of the output statistics to the parameters is assessed by calculating partial rank correlation coefficients between each parameter and each of the output statistics. These coefficients have the advantages of being nonparametric and of adjusting for the variation in the other parameters (23).
The distributions of the epidemic parameters (v, p, μ, μT, and φ) were taken directly from Blower et al. (7) with the following exceptions. We have a constant population size, N, which we select from a uniform distribution ranging from 1,000 to 1,000,000. R0,0 was drawn from a uniform distribution in the domain 1–10 (7), and as stated in the previous section, β0 was determined by the functional relationship between R0,0, β0, and the other parameters (Eq. 3).
The transposition parameters (ω1, γ, h, and c) were given broad ranges and specified as being uniformly distributed, reflecting the preliminary nature of the empirical data. The fraction of events leading to an additional copy γ was restricted to the range 0.4 to 0.6 because if γ is set too high, the artificial boundary produces spurious effects. Extremely low values of γ are biologically uninteresting (the element would rapidly be lost). The distributions of all parameters are shown in Table 1. Some of these parameters have triangular distributions, namely, their probability density functions are determined by linear segments. That is, the density function is
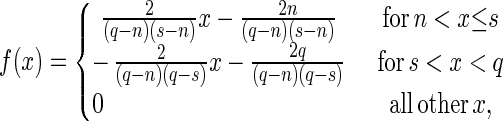 |
6 |
where n, s, and q are specified by Minimum, Peak, and Maximum, respectively in Table 1.
Table 1.
Distributions of parameters used for Latin hypercube sampling
| Parameter | Description | Minimum | Peak | Maximum |
|---|---|---|---|---|
| i0 | Initial copy number | 1 | 30 | |
| N | Population size | 1,000 | 1,000,000 | |
| R0,0 | Basic reproductive value | 1 | 10 | |
| ν | Progression rate | 0.0026 | 0.0053 | |
| p | Immediate progression | 0 | 0.05 | 0.3 |
| μ | Nondisease death rate | 0.0133 | 0.04 | |
| μT | Disease death rate | 0.0580 | 0.139 | 0.461 |
| φ | Recovery rate | 0.021 | 0.058 | 0.086 |
| h | Horizontal transfer rate | 0 | 0.001 | |
| ω1 | Transposition rate | 0.005 | 0.02 | |
| γ | Copy increase | 0.4 | 0.65 | |
| c | Cost per element | 0 | 0.002 |
The parameters i0 and N were chosen from discrete uniform distributions; p, μT, and φ had triangular distributions (described in text: see Eq. 6); the remainder were chosen from continuous uniform distributions.
The output statistics are calculated at equilibrium. These are (i) the mean copy number of the distribution, (ii) the variance in copy number, and (iii) the fraction f0 of active cases in which the strain involved is lacking IS elements. With 1,000 simulations, the partial rank correlation coefficients between the parameters and these statistics are given in Table 2. To test these correlations statistically, we calculate the critical partial rank correlation coefficients values beyond which the tabulated coefficients are significantly different from zero. With a Bonferroni correction for 36 tests, the critical values are ±0.0257 for a significance level of 0.01, and ±0.0241 for 0.001. We remark that these thresholds are only approximate because the structure of correlations among the input and output parameters is unknown.
Table 2.
Partial rank correlation coefficients between input parameters and output statistics
| Parameter | Mean | Variance | f0 |
|---|---|---|---|
| i0 | 0.033 | 0.046 | 0.017 |
| N | −0.008 | −0.012 | 0.020 |
| R0,0 | −0.408 | −0.420 | 0.270 |
| ν | −0.004 | −0.004 | 0.000 |
| p | −0.184 | −0.178 | 0.127 |
| μ | −0.222 | −0.258 | 0.118 |
| μT | −0.021 | −0.035 | 0.015 |
| φ | 0.014 | 0.009 | −0.009 |
| h | 0.736 | 0.560 | −0.890 |
| ω1 | −0.356 | −0.036 | 0.774 |
| γ | 0.864 | 0.900 | −0.673 |
| c | −0.505 | −0.525 | 0.383 |
The long run mean of the copy number distribution is very low, verifying the generality of the single simulation run of the previous section (Fig. 2). The median of mean copy numbers among the 1,000 runs was 1.44. The 10% and 90% quantiles were, respectively, 0.16 and 14.66.
It is evident that the epidemic parameters have a weak effect on all of the output variables. In contrast, the transposition parameters have a strong effect. The fraction of transpositions leading to an increase in copy number (γ) and horizontal transfer (h) increase the mean and variance and decrease the fraction of cases associated with the zero-copy strain. Selection against the element has exactly the opposite effect. While the transposition rate ω1 contributes both to increase and decrease in copy number, it counters the effect h at the zero-one copy boundary. Thus, it has roughly the opposite effect to h and γ on the output statistics.
The forces most clearly controlling changes in the copy number distribution are those associated with transposition, giving rise to a balance between horizontal transfer and replication on one hand, with selection and deletion on the other. Therefore, it is expected that the transposition parameters should have stronger influences than the epidemic parameters on the copy number distribution, and the extent of this effect is revealed by the Latin hypercube sampling/partial rank correlation coefficient analysis (Table 2). It is, however, surprising that μ and p should have such pronounced effects, and this should be investigated further. The large role played by horizontal transfer in the persistence of IS elements also suggests a future direction of inquiry in which the process is modeled in a more realistic fashion.
Numerical Investigation: Stochastic Simulation.
This section describes the basis for a Monte Carlo computer simulation of the model. We model the process in discrete time steps. Each individual may undergo six kinds of events in each time step: death caused by the disease, death from other causes, disease transmission, progression to the diseased state, recovery, and mutation of the strain responsible for infection. Whether or not each type of event has occurred is decided by a separate Bernoulli trial.
If the individual is susceptible, he or she may be infected with probability βiTi by another host infected with a strain carrying i copies. The time step length was kept small enough to ensure that β0N, which is the largest value the probability βiTi can take, was less than unity. With probability p, a new case will proceed directly to the active disease state, and otherwise enter the latent state. If an individual is latently infected, he or she may progress to the infectious state with probability v. The strain carried by individuals in the latent or active state may undergo change by one copy, with probabilities bi up or ai down. All individuals die with probability μ. Individuals with active TB, also may die from the disease, with probability μT. Recovery occurs with probability φ per person. We ran the simulation a total of 5,000 times, using the same parameter values as above, except with five different values of γ, the proportion of transposition events leading to an increase in copy number.
As in traditional population genetic models, the stochasticity produced by this process is important when N is small. Four kinds of outcomes are possible from this process. First, the IS goes to extinction, as anticipated from the deterministic model. Second, the epidemic never establishes itself because early stochastic fluctuations eliminate the parasite. Third, the epidemic establishes itself, but the entire distribution shifts to high copy numbers, with low-copy strains being lost by chance. The epidemic then consists of low-fitness strains (with copy numbers near or below iT) and eventually is extinguished. Notice that this outcome is possible in the stochastic but not in the deterministic model. Finally, if N is large, long-term persistence of IS elements (along with TB) is possible (results not shown).
Table 3 shows the results of this simulation, with different values of γ and illustrates the strong effect of this parameter on the extinction of M. tuberculosis along with the IS elements.
Table 3.
Outcomes from stochastic simulations
| γ | IS extinct | TB & IS extinct | Early extinction |
|---|---|---|---|
| 0.50 | 127 | 392 | 481 |
| 0.55 | 49 | 443 | 508 |
| 0.60 | 6 | 482 | 512 |
| 0.65 | 0 | 494 | 506 |
| 0.75 | 0 | 503 | 497 |
For each value of γ, 1,000 simulations were run. The population size is N = 1,000. Early extinction is defined as the extinction of TB before 20 years.
Thus, a stochastic framework leads to qualitatively different outcomes from those of the deterministic model. From these results, we predict that in sufficiently small and isolated human populations, drift-like effects may bring about the loss of IS elements, or both tuberculosis and IS, with probabilities that depend on the parameters. The sample described in the next section is drawn from an effectively large population that is probably exempt from such effects.
Observed Distributions
Fig. 3 shows the distribution of the number of bands in each DNA fingerprint of M. tuberculosis collected from San Francisco patients in the period 1991 to 1997 (ref. 2 and unpublished data). There is striking bimodality in this distribution.
Figure 3.

Overall distribution of band (copy) number from the San Francisco data set.
Subsets of this data set were taken based on country of birth; we present the four largest subsets (Fig. 4): United States (531 cases), China (224 cases), Philippines (204 cases), and Vietnam (79 cases). The “locations” (medians) of the distributions are clearly different when divided this way; this effect can be verified by using a nonparametric test based on ranks (Kruskal-Wallis; P ≪ 0.0001). The USA-born distribution is similar to the overall distribution, especially in having two modes. There is likely to be further heterogeneity within the USA-born cases.
Figure 4.

Copy number distributions of subgroups of the data set. The data were divided by place of birth of cases. We chose to plot the four categories with the largest totals: United States (open bars), China (dark gray bars), Philippines (light gray bars), and Vietnam (black bars).
By comparing the observed distribution (Fig. 3) with outcomes of the deterministic model (see previous), we suggest that the process is not at equilibrium and has multiple origins. We explore these possibilities next.
Discussion
We can view the entire process as an interplay between the symbiotic dynamics of TB in humans and IS elements in M. tuberculosis. The time scales of the two processes differ. Although it is known that TB dynamics are very slow (7) and that the IS-induced mutation rate is high, the relative time scales of these processes are such that M. tuberculosis reaches endemic equilibrium relatively quickly, after which the IS-based fingerprints continue to diversify. We caution that our model does not consider the details of the transmission or within-host processes, such as stochastic effects brought about by bottlenecks in the bacterial population, which may alter the speed at which equilibrium is approached. For a more detailed characterization of the process, further modeling and parameter estimation is necessary.
The highly skewed distribution at equilibrium in the deterministic case may lead to the extinction of the IS element in the stochastic case, as demonstrated in the Monte Carlo simulation. Note the similarity to the stochastic Wright-Fisher process in population genetics, where the zero-copy class, like extinction, is an absorbing state (in the absence of horizontal transfer in our case and mutation in population genetics). That the observed distribution (Fig. 3) is not heavily skewed and does not include many strains lacking IS elements (24) leads us to suggest that the observed IS copy number distribution is not at equilibrium. The peaks in the copy number distribution reflect recent events in the history of TB. There is evidence that M. tuberculosis is itself a relatively recent infection in humans (25). The interpretation that equilibrium has not been reached is also consistent with studies using secondary genetic markers in TB. Although it has been shown that secondary markers allow IS fingerprint clusters to be further differentiated, it is also evident that the states of these markers are correlated with IS genotypes (25–27).
Outbreaks, which are out of equilibrium, are expected generally to possess peaked distributions of repeated elements. That is, we predict that in isolated recently infected populations, especially those in which progression to disease is rapid, such as when HIV is prevalent, the copy-number distribution will be sharply peaked. It also may be fruitful to use this kind of genotyping (possibly with other markers) to estimate the time since an outbreak began.
In evolutionary time scales, the presence and absence of IS elements may be dynamic. The loss of a family of IS elements from a taxonomic group may be countered by the gain of other IS elements from distantly related taxa. Evidence has been uncovered for the occasional movement of transposable elements across taxonomic boundaries in the M. tuberculosis complex (28). Horizontal transfer of transposons also has been well studied in Drosophila species (29) and across a wider range of arthropods (30).
Sources of Bimodality.
The model presented here provides only a single (transient) mode apart from those at zero copies or at the artificial upper boundary. That the observed data show two modes and can be divided into separate groups that are statistically distinct suggests that some kind of heterogeneity is present. This heterogeneity may be spatial, temporal, behavioral/demographic, genetic, or a combination of such factors. There could be outbreaks initiated at separate times or locations. Each of these suboutbreaks, if it establishes itself in a population, will produce a different peak in copy number, and the overall distribution will exhibit multiple modes. The positions of the local peaks strongly depend on the initial copy number that was sampled. The host population might be demographically heterogeneous. Alternatively, or in addition, there could be genetically distinct strains of M. tuberculosis. If a high-fitness strain arises, for example through drug resistance, then another center of diversification is born that overrides the original fitness scheme laid out by the βi function and produces a separate peak that again depends on the copy number associated with the origin of the outbreak.
Persistence of IS.
While the extinction of the IS elements is one possible fate, there are two others to consider. First, the elements may persist stably or persist for a very long time until extinction. One potential source of persistence is horizontal transfer of the element, which if frequent enough, may stave off extinction. Although our model of this force is simplistic, the preliminary conclusion is that horizontal transfer in TB is too rare for it to increase the mean copy numbers to observed levels (21, 22) [for discussion of low conjugation rates in E. coli see Condit (31), Condit et al. (32), and Levin and Lenski (33)]. Therefore, in the case of IS6110, the answer to the problem of persistence lies elsewhere. Second, it may be that at least one copy of IS confers an advantage over none, as might be the case if there is coding information carried on the element. In this case, the copy number with the highest fitness (and frequency at equilbrium) would be near one and the element would still be somewhat prone to extinction. This possibility cannot, at equilibrium, account for the observed distribution. Third, persistence might be prolonged if the transposition rate is substantially higher than deletion rate (γ ≪ 0.5), and the element is regulated, that is, if the rate of transposition or deletion per element decreases as the copy number per genome rises. Fourth, the effects of IS element insertion around the genome are likely to be heterogeneous; in other words, there may be position effects. Strains with the least deleterious insertions will be selected. High-copy strains then may sidestep the fitness scheme of the process outlined here.
Finally, there may be occasional positive selection for strains carrying the element through direct or indirect fitness benefits to the host M. tuberculosis (compare ref. 34, a study using E. coli). In evolutionary time, even rare mutations conferring selective advantages to M. tuberculosis, linked to strains of intermediate copy number, may prevent the process from ever reaching equilibrium, thereby allowing long-term persistence of the IS element. IS elements can be viewed as “mutators,” the population theory of which has been studied (35–37). This explanation of IS persistence presupposes that the elements are abundant when a new mutation appears: selective sweeps occur frequently enough that the copy number distribution is never allowed to go to equilibrium. Even with this model, a family of IS elements occasionally will be lost. It is useful to remember that the particular families of IS elements chosen for characterization are those with high copy numbers (for the variability they produce) and it is possible that other families of IS have gone extinct.
Extinction of IS and TB.
The second alternative outcome occurs when the IS elements increase at a high rate (γ ≫ 0.5). If there is some cost to carrying the element, IS elements cause the epidemic to become extinguished. With a rapid increase in copy number, most strains will have more copies than the threshold copy number (iT) (for which R0,i = 1), and these strains will tend to be lost quickly. We predict this to occur in relatively small local host populations.
It may be advantageous for a transposable element to have comparable deletion and transposition rates. This principle should apply wherever there is a possibility of the host population being eliminated (i.e., when hard selection operates), such as in the case of infectious agents. However, we do not necessarily expect this characteristic when transposition is strongly regulated. While the preliminary estimate of γ used in our deterministic model is consistent with this hypothesis, it is important to obtain better estimates of this parameter.
Conclusions
We have proposed a dynamic model that combines both transposition of a genetic marker within a pathogen and the epidemiology of that parasite in the host population. Putting these kinds of processes together demonstrates the relative importance of each in shaping the distribution of copy number of the marker (IS6110). The epidemic parameters individually have weak effects. The overall influence of these parameters is indicated by the basic reproductive value, R0,0, which showed a stronger correlation with properties of the copy number distribution. In contrast, the transposition-related parameters have very strong effects on copy number distribution.
Clearly, the observed distribution of IS copy number is not described by the equilibrium distribution provided by the model. The bimodality of the empirical distribution reveals that the TB/IS system is heterogeneous and not at equilibrium. The data are effectively explained as a superposition of several transient episodes of the kind described by the model.
Acknowledgments
We thank L. Ancel, C. Bergstrom, S. Blower, B. Kerr, M. Lachmann, J. Pritchard, S. Ptak, J. Rhee, and N. Rosenberg for helpful discussions and comments. We also thank B. R. Levin and an anonymous reviewer. M.M.T. is grateful to the Research and Scholarships Office at the University of Sydney. This work is supported in part by National Institutes of Health Grants GM28016 and GM 28428 to M.W.F. and in part by National Institutes of Health Grant AI40906 to P.M.S.
Abbreviations
- IS
insertion sequence
- TB
tuberculosis
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.060564997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.060564997
References
- 1.Paul R E L, Packer M J, Walmsley M, Lagog M, Ranford-Cartwright L C, Paru R, Day K P. Science. 1995;269:1709–1711. doi: 10.1126/science.7569897. [DOI] [PubMed] [Google Scholar]
- 2.Small P M, Hopewell P C, Singh S P, Paz A, Parsonnet J, Ruston D C, Schecter G F, Daley C L, Schoolnik G K. N Engl J Med. 1994;330:1703–1709. doi: 10.1056/NEJM199406163302402. [DOI] [PubMed] [Google Scholar]
- 3.Small P M, Shafer R W, Hopewell P C, Singh S P, Murphy M J, Desmond E, Sierra M F, Schoolnik G K. N Engl J Med. 1993;328:1137–1144. doi: 10.1056/NEJM199304223281601. [DOI] [PubMed] [Google Scholar]
- 4.Daley C L, Small P M, Schecter G F, Schoolnik G K, McAdam R A, Jacobs W R, Hopewell P C. N Engl J Med. 1992;326:231–235. doi: 10.1056/NEJM199201233260404. [DOI] [PubMed] [Google Scholar]
- 5.Porco T C, Blower S. Theor Popul Biol. 1998;54:117–132. doi: 10.1006/tpbi.1998.1366. [DOI] [PubMed] [Google Scholar]
- 6.Blower S M, Small P M, Hopewell P C. Science. 1996;273:497–500. doi: 10.1126/science.273.5274.497. [DOI] [PubMed] [Google Scholar]
- 7.Blower S M, MacLean A R, Porco T C, Small P M, Hopewell P C, Sanchez M A, Moss A R. Nat Med. 1995;1:815–821. doi: 10.1038/nm0895-815. [DOI] [PubMed] [Google Scholar]
- 8.Ohta T, Kimura M. Proc Natl Acad Sci USA. 1981;78:1129–1132. doi: 10.1073/pnas.78.2.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brookfield J F Y. J Theor Biol. 1982;94:281–299. doi: 10.1016/0022-5193(82)90313-7. [DOI] [PubMed] [Google Scholar]
- 10.Charlesworth B, Charlesworth D. Genet Res. 1983;42:1–27. [Google Scholar]
- 11.Sawyer S, Hartl D. Theor Popul Biol. 1986;30:1–16. doi: 10.1016/0040-5809(86)90021-3. [DOI] [PubMed] [Google Scholar]
- 12.Moody M E. J Math Biol. 1988;26:347–357. doi: 10.1007/BF00277395. [DOI] [PubMed] [Google Scholar]
- 13.Ohta T, Kimura M. Genet Res. 1973;22:201–204. doi: 10.1017/s0016672300012994. [DOI] [PubMed] [Google Scholar]
- 14.Moran P A P. Theor Popul Biol. 1975;8:318–330. doi: 10.1016/0040-5809(75)90049-0. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein D B, Ruiz Linares A, Cavalli-Sforza L L, Feldman M W. Genetics. 1995;139:463–471. doi: 10.1093/genetics/139.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhivotovsky L A, Feldman M W. Proc Natl Acad Sci USA. 1995;92:11549–11552. doi: 10.1073/pnas.92.25.11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldman M W, Bergman A, Pollock D D, Goldstein D B. Genetics. 1997;145:207–216. doi: 10.1093/genetics/145.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh R W, de Leon A P, Agasino C B, Hahn J A, Daley C L, Hopewell P C, Small P M. J Infect Dis. 1998;177:1107–1111. doi: 10.1086/517406. [DOI] [PubMed] [Google Scholar]
- 19.Niemann S, Richter E, Rusch-Gerdes S. J Clin Microbiol. 1999;37:409–412. doi: 10.1128/jcm.37.2.409-412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naas T, Blot M, Fitch W M, Arber W. Mol Biol Evol. 1995;12:198–207. doi: 10.1093/oxfordjournals.molbev.a040198. [DOI] [PubMed] [Google Scholar]
- 21.McFadden J. Mol Microbiol. 1996;21:205–211. doi: 10.1046/j.1365-2958.1996.6271345.x. [DOI] [PubMed] [Google Scholar]
- 22.Balasubramanian V, Pavelka M S, Jr, Bardarov S S, Martin J, Weisbrod T R, McAdam R A, Bloom B R, Jacobs W R., Jr J Bacteriol. 1996;178:273–279. doi: 10.1128/jb.178.1.273-279.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blower S M, Dowlatabadi H. Int Statist Rev. 1994;62:229–243. [Google Scholar]
- 24.Agasino C B, de Leon A P, Jasmer R M, Small P M. Int J Tuberculosis Lung Dis. 1998;2:518–520. [PubMed] [Google Scholar]
- 25.Sreevatsan S, Pan X, Stockbauer K E, Connell N D, Kreiswirth B N, Whittam T S, Musser J M. Proc Natl Acad Sci USA. 1997;94:9869–9874. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burman W J, Reves R R, Hawkes A P, Rietmeijer C A, Yang Z, El-Hajj H, Bates J H, Cave M D. Am J Respir Crit Care Med. 1997;155:1140–1146. doi: 10.1164/ajrccm.155.3.9117000. [DOI] [PubMed] [Google Scholar]
- 27.van Soolingen D, de Haas P E W, Hermans P W M, Groenen P M A, van Embden J D A. J Clin Microbiol. 1993;31:1987–1995. doi: 10.1128/jcm.31.8.1987-1995.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mariani F, Piccolella E, Colizzi V, Rappuoli R, Gross R. J Gen Microbiol. 1993;139:1767–1772. doi: 10.1099/00221287-139-8-1767. [DOI] [PubMed] [Google Scholar]
- 29.Kidwell M G. Genetica. 1992;86:275–286. doi: 10.1007/BF00133726. [DOI] [PubMed] [Google Scholar]
- 30.Robertson H M. Nature (London) 1993;362:241–245. doi: 10.1038/362241a0. [DOI] [PubMed] [Google Scholar]
- 31.Condit R. Evolution. 1990;44:347–359. doi: 10.1111/j.1558-5646.1990.tb05204.x. [DOI] [PubMed] [Google Scholar]
- 32.Condit R, Stewart F M, Levin B R. Am Nat. 1988;132:129–147. [Google Scholar]
- 33.Levin B R, Lenski R E. In: Coevolution. Futuyma D J, Slatkin M, editors. Sunderland, MA: Sinauer; 1983. pp. 99–127. [Google Scholar]
- 34.Chao L, Vargas C, Spear B B, Cox E C. Nature (London) 1983;303:633–635. doi: 10.1038/303633a0. [DOI] [PubMed] [Google Scholar]
- 35.Liberman U, Feldman M W. Theor Popul Biol. 1986;30:125–142. doi: 10.1016/0040-5809(86)90028-6. [DOI] [PubMed] [Google Scholar]
- 36.Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon P H, Godelle B. Nature (London) 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- 37.Tenaillon O, Toupance B, Le Nagard H, Taddei F, Godelle B. Genetics. 1999;152:485–493. doi: 10.1093/genetics/152.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]