

Abstract
A number of theories propose that RNA, or an RNA-like substance, played a role in the origin of life. Usually, such hypotheses presume that the Watson–Crick bases were readily available on prebiotic Earth, for spontaneous incorporation into a replicator. Cytosine, however, has not been reported in analyses of meteorites nor is it among the products of electric spark discharge experiments. The reported prebiotic syntheses of cytosine involve the reaction of cyanoacetylene (or its hydrolysis product, cyanoacetaldehyde), with cyanate, cyanogen, or urea. These substances undergo side reactions with common nucleophiles that appear to proceed more rapidly than cytosine formation. To favor cytosine formation, reactant concentrations are required that are implausible in a natural setting. Furthermore, cytosine is consumed by deamination (the half-life for deamination at 25°C is ≈340 yr) and other reactions. No reactions have been described thus far that would produce cytosine, even in a specialized local setting, at a rate sufficient to compensate for its decomposition. On the basis of this evidence, it appears quite unlikely that cytosine played a role in the origin of life. Theories that involve replicators that function without the Watson–Crick pairs, or no replicator at all, remain as viable alternatives.
Among the most commonly encountered ideas concerning the origin of life is the one that it involved an “RNA world” at an early stage (1). The term was coined by Gilbert (2), who also stated “The first stage of evolution proceeeds, then, by RNA molecules performing the catalytic activities necessary to assemble themselves out of a nucleotide soup.” The existence of such a soup has generally been taken for granted. For example, Eigen and Schuster (3) wrote “The building blocks of polynucleotides—the four bases, ribose and phosphate were available too under prebiotic conditions. Material was available from steadily refilling pools for the formation of polymers, among them polypeptides and polynucleotides.” The experimental evidence to date, however, does not appear to support such claims.
Many problems have arisen with both the prebiotic synthesis and the stability of ribose (4–9). To avoid the need for ribose, some authors have preferred to invoke an RNA-like polymer, with a simpler or more accessible backbone, at the start of life (6, 10–16). A pre-RNA world would have come first, during which some substance of this type carried out the genetic functions later taken over by RNA. In the great majority of these theories, Watson–Crick pairing of A with U and of G with C is retained as the basis of genetic template recognition.
These suggestions still presume that the bases adenine, cytosine, guanine, and uracil were readily available on early Earth. I have argued that this presumption is not supported by the existing knowledge of the basic chemistry of these substances (4, 17). If the availability of the Watson–Crick pairs at the start of life appears implausible, then more attention must be given to theories that employ a very different replicator or no replicator at all.
To provide a firm basis for this conclusion, I have undertaken a series of reviews in which I consider in detail the chemical evidence for the availability of the Watson–Crick bases at the start of life. In a previous paper, however, I concluded that current information concerning the availability and chemical properties of adenine did not support the idea that it was used in a replicator at the start of life (17). In this publication, I wish to consider the prebiotic syntheses and the stability of cytosine.
RESULTS AND DISCUSSION
Absence of Cytosine in Meteorites and Electrical Spark Discharge Experiments.
The isolation of adenine and guanine from meteorites has been cited as evidence that these substances might have been available as “raw material” on prebiotic Earth (18). However, acid hydrolyses have been needed to release these materials, and the amounts isolated have been low (17–19). Traces of uracil have also been reported in such analyses (20), but no cytosine at all.
The formation of a substance in an electric spark discharge conducted in a simulated early atmosphere has also been regarded as a positive indication of its prebiotic availability (21). Again, low yields of adenine and guanine have been reported in such reactions, but no cytosine (22). The failure to isolate even traces of cytosine in these procedures signals the presence of some problem with its synthesis and/or stability.
Proposed Prebiotic Cytosine Syntheses.
As bonds from carbon to a hetero atom are more readily constructed than carbon–carbon bonds, cytosine syntheses have usually combined a three-carbon fragment with another bearing a urea-like carbon. The most prominent C-3 fragments used have been cyanoacetylene and its hydrolysis product, cyanoacetaldehyde. These processes are discussed separately below.
Syntheses based on cyanoacetylene.
As shown in Fig. 1(Fig. 1), Ferris et al. (23) reported that 0.2 M cyanoacetylene (I) and 2 M cyanate (II) reacted together readily at 30°C to give trans-cyanovinylurea (III) and unidentified products. Conversion of trans-cyanovinylurea to cytosine (with the cis isomer as a likely intermediate) took place readily at pH 11 or greater. In a more direct preparation, cyanate and cyanoacetylene were heated together at 100°C for 24 hr. In a typical run at low concentration, 0.025 M cyanoacetylene and 0.05 M cyanate (the stoichiometry requires two cyanates per cyanoacetylene) afforded 6% cytosine. The maximum yield observed over all circumstances was 19%.
Figure 1.

Principal proposed prebiotic routes to cytosine. The hydrolysis products of the reactants and of cytosine are included in the scheme.
Questions arise, however, concerning the availability of the reactants on early Earth. Cyanoacetylene can be produced in a spark discharge in a CH4/N2 mixture as the second most prevalent product (up to a maximum of 8.4% of the principal product, HCN) (23, 24). This mixture, which introduces carbon in reduced form but excludes ammonia and water, is an unlikely candidate for Earth’s atmosphere at the time of the origin of life. That atmosphere was “… probably dominated by CO2 and N2, with traces of CO, H2, and reduced sulfur gases”, unless a volcanic source of methane and ammonia was present (25). By contrast, when ammonia (24) or hydrogen sulfide (26) are included in spark discharge experiments, little cyanoacetylene is produced. The aspartic acid and asparagine that are formed under those conditions arise to some extent from the reaction of cyanoacetylene with HCN and ammonia (27).
An extensive solution chemistry for cyanoacetylene with simple nucleophiles has been recorded. Ammonia (28), amines (29–30), thiols (31), HCN (32), and “the commonly used alkaline buffers” (23), react rapidly at room temperature or lower with cyanoacetylene. In certain cases, e.g., the reaction of cyanoacetylene with phosphate, the product hydrolyzes to afford cyanoacetaldehyde (Fig. 1, IV) (33). But for many reactions of cyanoacetylene, the products appear stable or react further to afford further transformation products. The rates of reaction of cyanoacetylene with simple nucleophiles appear more rapid than its reaction with cyanate, but no direct competition experiments have been reported. In the absence of other nucleophiles, cyanoacetylene is hydrolyzed by water to cyanoacetaldehyde. The half-life at pH 9 and 30°C has been estimated as about 11 days (0.03 yr) (34).
The prebiotic availability of cyanate also is undetermined. It can be produced by hydrolysis of cyanogen (23), a simple substance that may be widely distributed elsewhere in the universe, as it has been detected in the atmosphere of Titan (35), and related nitriles are prominent components of interstellar clouds (36). No simulations have been carried out to estimate the formation of cyanogen under plausible early Earth conditions, however. Cyanogen’s stability in aqueous solutions is limited because it is decomposed by both base and acid. At 25°C and pH 9 (the usual pH for HCN oligomerization), cyanogen’s half-life can be estimated from existing data (37) to be less than 30 sec. Cyanogen or cyanoformamide (an intermediate hydrolysis product of cyanogen) can replace cyanate in its reaction with cyanoacetylene, affording cyanovinylurea at lower yields. At pH 8 and room temperature (9 days reaction time, 0.1 M reactants), yields of cyanovinylurea were 2.9% from cyanoformamide, 3.0% from cyanogen, and 4.7% from cyanate.
Small amounts of ammonium cyanate also exist in equilibrium with urea (ref. 38, and see below) but these quantities would appear insufficient for the synthesis. A reaction of 1 M urea with 1 M cyanoacetylene (100°C, 20 hr) afforded 4.8% cytosine, but no cytosine was detected when reagent concentrations were reduced to 0.1 M (23). Much higher concentrations of urea than of cyanate are needed for cytosine synthesis. Urea might have been more available than cyanate on prebiotic Earth, however, as its formation in electric discharge experiments under an atmosphere of N2, CO, and H2O has been reported (39).
Apart from the questions concerning its availability, cyanate is unstable in aqueous solution, hydrolyzing to ammonium carbonate with a half-life of “… at most a hundred years” (23). For these reasons, the authors concluded that “it is difficult to see how concentrations (of cyanate and cyanoacetylene) as high as 0.01 M could have accumulated in the primitive ocean.”
Syntheses based on cyanoacetaldehyde.
The synthesis of cytosine from cyanoacetaldehyde (Fig. 1, IV) and urea (V) was reported by Robertson and Miller (40). The authors described this reaction in the following terms. “Here we show that in concentrated urea solution—such as might have been found in evaporating lagoons or in pools on drying beaches on early Earth—cyanoacetaldehyde reacts to form cytosine in yields of 30–50%, from which uracil can be formed by hydrolysis… The previous lack of plausible prebiotic syntheses of cytosine and uracil has led some other authors to suggest that other bases were used in the first genetic material. These reactions provide a plausible route to the pyrimidine bases required in the RNA world.” This reaction is closely related to the synthesis of cytosine from cyanoacetylene and urea (23) described above. Robertson and Miller (40) noted this in a correction: “Replacing cyanoacetylene by its hydrolysis product cyanoacetaldehyde gives essentially the same yield, suggesting that the cyanoacetylene reaction may have gone through cyanoacetaldehyde.”
The Robertson-Miller procedure was conducted by heating 10−3 M cyanoacetaldehyde with varying urea concentrations (expressed as molality) in a sealed ampule at 100°C. For kinetic purposes, the reaction was stopped after 5 hr. The rate equation contained first-order and second-order terms in urea. The half-life in 1 molal urea can be calculated from their equation as 180 hr. In 10 molal urea, it is 67 hr. The maximum yield was determined from much longer runs and expressed as the sum of uracil (formed by deamination) and cytosine. This sum rose sharply with urea concentration at low concentration (about 20% in 5 molal urea) and then leveled off to about 33% in 20 molal urea. The yield of cytosine alone, reported only for a reaction run in saturated urea (120 molal), was 53%. An Arrhenius plot of their data gave a heat of activation of 28.2 kcal (1 cal = 4.18 J). At 25°C, the half-life would be 300 yr with 1 M urea and 15 yr with 20 molal urea (the maximum concentration attainable at that temperature).
An obvious difficulty with this reaction is that the formation of cytosine and the subsequent deamination of the product to uracil (see below) occur at the about the same rate when urea concentrations are 1–2 M. Robertson and Miller (40) noted that 40% of cytosine was deaminated after 120 hr at 100°C and that in saturated urea, cytosine yields fell after 30 hr because of deamination. It is clear that the yield of cytosine would fall to 0% if the reaction were extended for a number of half-lives. This provides no difficulty in the laboratory, where one can start with a urea concentration of one’s choice and monitor the time carefully. On early Earth, the following circumstances would be needed: An isolated lagoon or other body of sea water would have to undergo extreme concentration, to perhaps 10−5 of its initial volume. This reduction in volume would be needed to bring urea from a concentration of 10−4 to 10−5 M assumed for many substances in a prebiotic ocean (see below) to that necessary for the reaction. It would further be necessary that the residual liquid be held in an impermeable vessel, for reasons described below. The concentration process would have to be interrupted for some decades (assuming a temperature near 25°C) with the urea concentration near saturation, to allow the reaction to occur. At this point, the reaction would require quenching (perhaps by evaporation to dryness) to prevent loss by deamination. At the end, one would have a batch of urea in solid form, containing some cytosine (and uracil). This sequence cannot be excluded as a rare event on early Earth, but it cannot be termed plausible.
The above circumstances do not provide the only barrier to the success of the reaction. Questions arise about the availability of cyanoacetaldehyde. Browne (41) termed it “… another quite common component of sea water that owes its formation partly to lightning bolts.” Robertson and Miller (40) justify its prebiotic availability as a hydrolysis product of cyanoacetylene, which in turn is “… produced from a spark discharge in a CH4/N2 mixture [23, 24] and is an abundant interstellar molecule.” As we have explained above, however, the questionable availability of cyanoacetylene and its reactivity to a broad variety of common nucleophiles make it an unreliable source for cyanoacetaldehyde.
Cyanoacetaldehyde also is vulnerable to reaction with a number of chemicals considered to be prebiotic. Its reaction with cyanide has been described theoretically (42), although it apparently has not carried out experimentally. Cyanoacetaldehyde is in equilibrium with a dimer (23), which then reacts readily with thiols (43). Its reaction with amino groups of proteins (44) suggests that simple amino acids will also combine with it. The great chemical activity exhibited by both cyanoacetaldehyde and its precursor suggest that little of either will persist in prebiotic media, even before the start of a concentration process. The synthesis of Robertson and Miller (40) was carried out with the exclusion other possible prebiotic chemicals that might have interfered. Furthermore, no experiments were reported to assess the effect of concentrated brine (expected from the evaporation of seawater) on the synthesis.
In the case of cyanoacetaldehyde, however, one disruptive chemical could not be excluded: water. Although it is more stable than its precursor, cyanoacetaldehyde is subject to hydrolysis to acetonitrile and formate, with a half-life of 31 yr at 30°C and pH 9 (34). This rate makes the reaction competitive with cytosine formation at the lower end of the range of urea concentrations studied (40). When a combination of 0.1 M urea and 0.1 M cyanoacetaldehyde were heated together earlier by other workers (34), no pyrimidines were detected. We deduced earlier that an extreme concentration process (to about 1:105) was necessary to bring urea to a concentration suitable for reaction with cyanoacetaldehyde. But unless that concentration took place very rapidly (years, not decades) on a geologic time scale, any initial cyanoacetaldehyde would be unlikely to survive the process.
Urea would also be at risk during a lengthy evaporative process. It exists in equilibrium with a small amount of ammonium cyanate (Ke at 60°C = 1.04 × 10−4) (38). This equilibrium will be shifted to the right continually by further hydrolysis of cyanate to carbonate and (in an open system) escape of ammonia. The cytosine synthesis of Robertson and Miller was carried out in a sealed tube, which prevented ammonia loss. The rate constant for urea decomposition at 60°C is 2.6 × 106⋅s−1 and the half-life is 30 days. The half-life at 25°C has been estimated at 25 yr (27). This is comparable with the rate of cytosine synthesis in concentrated urea solutions, but of course decomposition could take place during the concentration phase as well. Other substances likely to be present on early Earth could also consume the urea during the concentration process. When 0.1 M glycine and 0.1 M urea are heated together in a sealed tube at 100°C for 10 hr, >50% of the glycine is converted to N-carbamoyl glycine. The carbamoylating agent is cyanate, formed from the urea (45). When a similar reaction was run in an open system to facilitate ammonia loss, half of the urea was destroyed after 5 hr at 90°C and pH 7 (46). Diglycine, oligoglycines, and diketopiperazines were other reaction products. These reactions appear more rapid than cytosine synthesis and take place at lower urea concentrations. Unless amines and amino acids were excluded, they would presumably prevent cytosine synthesis.
The combination of circumstances described above limits the cytosine synthesis from urea and cyanoacetaldehyde to circumstances in which concentrated urea can be employed from the beginning, competing nucleophiles can be excluded, and the time can be controlled carefully.
Deamination of Cytosine.
As we saw in the previous section, the spontaneous deamination of cytosine and its derivatives in aqueous solutions provides an obstacle in prebiotic preparations of these substances. This reaction was first reported from our own laboratories (47–48). It takes place at a sufficient rate in single-stranded DNA for it to constitute a genetic hazard (49–50). Cells are normally protected from the reaction by the repair enzyme, uracil-DNA glycosylase, but in the absence of that enzyme, enhanced mutagenesis occurs (51).
The most detailed studies of the deamination of cytosine and cytidine have been carried out by Garrett and Tsou (52). They reported that the reaction occurs at all pH values but is minimal in the range 6–9. Acid catalyzes the reaction by protonation of cytosine, and base speeds it by direct attack on cytosine. A variety of buffers also catalyze the reaction (47, 52) with bisulfite having a particularly strong effect (53–54). The data of Garrett and Tsou have recently been extended by Levy and Miller (55), who estimated, by extrapolation, a half-life of 340 yr at pH 7 and 25°C for cytosine (uncatalyzed reaction). This corresponds to a rate of 6.5 × 10−11⋅s−1. This rate was increased by common buffers, for example, by 50% in 0.05 M acetate. The value is roughly compatible with those calculated by others for the deamination of cytidine and single-stranded DNA (49, 56–58). The activation energy reported in these studies is in the range of 26–29 kcal/mol.
This situation was summarized some years ago: “Cytosine hydrolyzes to uracil rather rapidly and cytidine is hydrolyzed to uridine at a similar rate… This is a real difficulty if it is assumed that cytosine was required for nucleic acids in the first organism.” (27).
Deamination, of course, is not the only hazard; other chemical reactions will also deplete cytosine supplies. For example, exposure to solar UV light on early Earth would quickly convert cytosine to its photohydrate and to cyclobutane photodimers (both very susceptible to deamination) (59). Such reactions would place an additional requirement on prebiotic cytosine syntheses: they must be carried out in the dark.
Prebiotic Plausibility of Cytosine Synthesis.
The assembly of a cytosine-containing replicator would require several steps beyond cytosine synthesis as well as the concurrent synthesis of the other replicator components. We wish to consider the suitability of the reactions described above for the prebiotic synthesis of cytosine, not in trace amounts but in the quantities needed to support further chemical transformations. We must assume that prebiotic cytosine synthesis took place at a rate that replaced the material lost by deamination, to maintain a steady-state concentration. The synthetic requirements depend on the assumptions made about the environment in which the replicator was assembled.
Synthesis in a global ocean.
This concept underlies the Oparin-Haldane hypothesis, a central paradigm of the origin-of-life field (see ref. 27 for a comprehensive list of references). In this account, the origin of life took place in such an ocean, sometimes termed “the prebiotic soup.” Possible concentrations for a number of substances such as adenine, ammonia, HCN, and total amino acids in a prebiotic ocean have been estimated and fall in the range from 10−5 to 10−4 M. (21, 60–61). Although an adenine concentration in this range has been considered as possibly too low for useful prebiotic synthesis (21), we will assume that a steady-state concentration of 10−5 M will suffice. We can then ask what rate of synthesis is needed to maintain a 10−5 M cytosine concentration in the soup?
If we use the data of Levy and Miller (55) at 25°C at pH 7, with dilute buffer catalysis (and excluding bisulfite), the rate of cytosine deamination is 10−10⋅s−1 × 10−5 M = 10−15 M⋅s−1. Cytosine has not been reported as a direct product of atmospheric processes, so we will assume that it is made by the reactions of two substances, A and B, that are produced globally and occur in the soup at a concentration of 10−5 M. The rate constant, Ksyn, for this reaction must be sufficient so that the cytosine produced balances that lost by deamination. (The synthetic reaction, of course, is second-order, whereas the deamination of cytosine is pseudo-first-order.) We then get
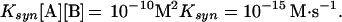 |
Solving to find the needed rate constant, we find that Ksyn = 10−5 M−1⋅s−1 at 25°C. A reaction with this rate constant should be readily demonstrable in the laboratory at 25°C. If A and B were at an initial concentration of 1 M, they should react to the extent of 10% in about 3 hr. Note that in this estimate, we have assumed neutral pH to minimize deamination. We have also ignored the possibility that A and B react with other substances present in the soup. In practice, the required Ksyn is likely to be higher. The reactions described above for prebiotic cytosine synthesis did not meet this requirement. They required reaction times of hours at 100°C. In the case of the cyanoacetaldehyde–urea combination, reaction at 25°C would require hundreds of years.
It has been argued that a cold or frozen condition for early Earth would be more favorable for the origin of life, as it would slow the decomposition of cytosine and the other bases (55). It is not obvious that any advantage would be gained by this, however, as the rate of the synthetic reaction would also be slowed on a frozen Earth. A careful study of the temperature profiles of the competing synthetic and degradative reactions would be needed to determine whether any significant advantage can be obtained by manipulating the reaction temperature.
Of course, some new combination of chemicals and an appropriate set of conditions may yet be encountered that would produce cytosine at the needed rate. This possibility seems unlikely, because the combinations of substances that may be present in the soup and react to form cytosine are limited and have already been explored to some extent.
The “drying lagoon” scenario.
This term was used by Robertson and Miller (40) to describe processes in which reactants are concentrated in specific geologic environments such as lagoons, thereby enhancing their reaction rates. Very different environments could be used to synthesize the different replicator components. After synthesis, the components would be released into the open ocean, where final assembly could take place. As the purine and pyrimidine components of a replicator would be prepared in different environments, the replicator synthesis would take place in the open ocean. [This idea was attributed to Robertson and Miller by Browne (41)].
In this scenario, the concentrations of A and B could be enhanced greatly by concentration in a drying lagoon or comparable locale. For example, if A and B were both at 1 M, the rate would be enhanced by 1010, and a smaller value for Ksyn might suffice. However, this gain in rate would be largely overcome by the effects of the subsequent dilution. Deamination would take place on a global scale, but synthesis only within a limited locale. The available evidence suggests that volcanic islands constituted the principal land areas on early Earth, with the continents much smaller than their present size (62). If this were the case, then much less shoreline would be available for lagoon formation. No firm data exists concerning the extent of the ocean and the distribution of drying lagoons on early Earth, however, so we will use current information for our estimate. The volume of the oceans at present is 1.3 × 1018 m3 (63). A typical large lagoon such as the Sivash (Crimea) or the McLeod Evaporite Basin (western Australia) may hold 2 × 109 m3 of water (64–65). If we compare the initial volume of the vessel in which synthesis is to be carried out with that of the one available for deamination, we see that the latter exceeds the former by 6 × 108. Thus, the dilution factor dissipates much of the advantage produced by concentration of reactants.
Another potent factor acts to penalize the synthesis, however. The lagoon process would operate in a batchwise manner, and require the following steps. (i) Formation of a barrier, to isolate the lagoon from the ocean. (ii) Evaporation of the lagoon to the optimal size for reaction. If we assume that an initial concentration of the reactants was 1 × 10−5 M, and one of 1 M was needed, then a concentration of the lagoon to 1/100,000 of its initial volume would be required. (iii) Cytosine synthesis. (iv) Rupture of the barrier, releasing cytosine into the sea.
Only the time spent in stage (iii) would be chemically productive; the remainder would serve to reduce the effective rate. The geological literature offers no evidence that the other steps occur with any great frequency:
Coastal lagoons are common on this planet today, but according to Barnes (64), “lagoons are rarely completely isolated from the sea. Characteristically, they have a channel (or series of channels) through which water is exchanged with the larger adjacent water body.” In cases where a lagoon becomes isolated, it usually evolves into a freshwater lake or pond. Alternatively, the ocean may rupture the barrier and reform a bay.
Commonly, rain, river water, aerosols from the sea, and seawater seepage replenish lagoons (65). In certain instances, however, a lagoon may evaporate. This occurs when evaporation exceeds infall through rain and various forms of inflow. “This condition is found in coastal zones of the semi-arid to arid belts that girdle the planet between latitudes 15 and 35°” (65). Evaporation can then range from 2 to 8 m/yr. A well defined order of salts is precipitated (evaporites), until the residual brines (bitterns) precipitate sodium chloride and magnesium and potassium salts when the volume has reached 5% of its initial value (65–66). However, as the density of the brines rises through evaporation, there is an increasing tendency for them to escape through seepage. Inflow of seawater also serves to reduce the density differential between the sea and the brines. The drop in vapor pressure with the increase in salt concentration also tends to limit evaporation. Evaporite systems normally approach a steady-state system in which finite volumes of brine remain. Complete evaporation rarely, if ever, occurs (66). If further volume reduction does take place, the brines sink to subsurface levels, where evaporative loss is suppressed. (65). These fluid inclusions (up to 10% by volume within halite) may persist through burial, until geological changes affect the bed (67). If today’s Earth may be taken as a model for the early one, then, cases of extreme lagoon concentration (to the extent needed to concentrate a solute by 105) are rare or nonexistent. This mechanism cannot be considered as a source that could stock a global ocean with a particular chemical.
Synthesis in a restricted location.
If the chemistry that started life is confined to a single location, then questions concerning global distribution of chemicals or the abundance of a particular location become unimportant. The origin of life can be seen as a unique event. Charles Darwin selected a “warm little pond” as the locale that he favored for the origin of life. Many other sites have been suggested, for example deep-sea thermal vents (68), comet ponds (69), and clouds (70).
Unlike the environments described above, a scenario of this type cannot be excluded by abstract chemical reasoning. In a publication concerning the cyanate-cyanoacetylene synthesis of cytosine, for example, the authors speculated that “Perhaps cyanate could have concentrated somewhat during the evaporation of pools and then reacted with cyanoacetylene from the atmosphere, but this mechanism is not very convincing” (23). They added: “the instability of cyanate and cyanoacetylene restricts severely the range of prebiotic environments in which such a synthesis could have occurred.”
Levy and Miller (55) raised the possibility that unknown concentration mechanisms could have raised the concentration of cytosine in an environment. Further chemical processes might then incorporate it into a hydrogen-bonded polymer, protecting it from deamination (For the protective effect of nucleic acid secondary structure on cytosine deamination, see refs. 49 and 71). If a sufficient number of unique geochemical environments were formed within proximity of one another, they might conceivably catalyze a number of chemical reactions which, taken in sequence, would serve to construct a replicator. This possibility has been illustrated by Arrhenius et al. (72).
A scenario of this type would be more credible for uracil than cytosine. Once synthesized by the cyanate-cyanoacetylene reaction, or by another process, uracil would persist for a considerably longer time than cytosine. The half-life for uracil decomposition by hydrolysis has been reported to be 12 yr at 100°C and 3.8 × 108 yr at 0°C (55). However, the restrictions imposed by the instability of cyanate and cyanoacetaldehyde would still apply. Furthermore, a number of specific catalyzed processes would still be needed to incorporate uracil into a replicator. With each of these steps, the fraction of the desired product in the mixture would diminish, and the amount of interfering side products would increase. Processes that would lead to the purification of the desired intermediate can undoubtedly be specified, but they would compete with numerous other natural processes that would have the reverse effect.
A series of productive steps culminating in the synthesis of a replicator can never be excluded, particularly when many of the key processes have not been demonstrated. We must assume that each of them, whether a reaction, a transfer, or a concentration, would have constraints that would limit its probability. The events are not linked in any way, as the process of classical natural selection would take over only when a functioning replicator had come into existence. The likelihood of the formation of such a replicator would then be the product of the probabilities of the various individual steps. If the replicator was complex, chemically, and the steps numerous, than an explanation of this type would portray the origin of life as a highly improbable event. This position has been captured by Jacques Monod in his book Chance and Necessity (73):
“… Life appeared on earth: what, before the event, were the chances that this would occur? The present structure of the biosphere far from excludes the possibility that the decisive event occurred only once. Which would mean its a priori probability was virtually zero… This idea is distasteful to many scientists. Science can neither say nor do anything about a unique occurrence… If it was unique, as may perhaps have been the appearance of life itself, then before it did appear its chances of doing so were infinitely slender. The universe was not pregnant with life nor the biosphere with man. Our number came up in the Monte Carlo game.”
As scientists, we have not yet been forced into this position. Alternatives remain yet for the origin of life that do not involve the difficulties of the chemistry of RNA-like substances.
CONCLUSIONS
The deamination of cytosine and its destruction by other processes such as photochemical reactions place severe constraints on prebiotic cytosine syntheses. If cytosine concentrations are to be maintained on a worldwide basis, then synthesis must be sufficient to replace depletion. The syntheses described thus far do not possess the necessary speed and selectivity to meet this requirement. The use of drying lagoons as a site for prebiotic synthesis has been suggested as a remedy: synthetic rates would be enhanced by greatly increasing the concentration of the reagents. The lagoon suggestion appears geologically implausible, however. All schemes in which cytosine is synthesized locally and distributed globally also are handicapped in that the enormous dilution that takes place when cytosine is released into a global sea offsets any gain in synthetic efficiency.
The possibility remains that a set of unique circumstances produced a batch of cytosine on one or a few occasions on early Earth. The cyanoacetylene-cyanate path seems the most likely candidate yet described for such an event, although problems remain concerning the availability of the reactants. Unless the cytosine produced were quickly processed, however, such an event would not be significant for the origin of life. Decomposition processes would gradually consume the product.
This fate would be avoided if the cytosine were used soon after it was made. The rate of cytosine deamination is not affected appreciably when it is part of a nucleotide or single-stranded nucleic acid, but it is slowed by a factor of 140 on incorporation into double-stranded DNA (58). Rapid incorporation of cytosine into a double-stranded replicator could best be achieved if all components (coding units and backbone) were synthesized under the same set of conditions, and polymer formation took place in the same environment. To avoid cytosine loss, this process should take at most several centuries at 25°C. A change in temperature would not improve matters unless it could be shown that the synthetic processes were retarded less, or enhanced more, than the degradative ones at a different temperature.
Suitable chemistry for such transformations has not been demonstrated, however, and may not exist. The evidence that is available at the present time does not support the idea that RNA, or an alternative replicator that uses the current set of RNA bases, was present at the start of life. This conclusion could be reversed if a prebiotic simulation were devised that produced all of the bases in good yield under a single set of conditions, by using a plausible combination of water, atmospheric components, and minerals. In the absence of such a demonstration, more attention should be given to origin-of-life theories that do not require the four RNA bases: (i) The first living system used a replicator constructed of more accessible and stable components. A number of possibilities may exist, with the clay system of A.G. Cairns-Smith (74) perhaps the best known. (ii) Life began with cycles of autocatalytic reactions. Storage and transfer of information at the polymer level came later. A number of writers have discussed this possibility, including F. Dyson (75) and S. Kauffman (76). One possible system has been described in detail by G. Wächtershäuser (77).
Acknowledgments
I am indebted to Dr. Leslie Orgel for his helpful suggestions concerning this manuscript.
References
- 1.Gesteland R F, Cech T R, Atkins R A, editors. The RNA World. 2nd Ed. Plainview, New York: Cold Spring Harbor Lab. Press; 1998. [Google Scholar]
- 2.Gilbert W. Nature (London) 1986;319:618. [Google Scholar]
- 3.Eigen M, Schuster P. J Mol Evol. 1982;19:47–61. doi: 10.1007/BF02100223. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro R. Origins Life. 1984;14:565–570. doi: 10.1007/BF00933705. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro R. Origins Life Evol Biosphere. 1988;18:71–85. doi: 10.1007/BF01808782. [DOI] [PubMed] [Google Scholar]
- 6.Joyce G F, Schwartz A W, Miller S L, Orgel L E. Proc Natl Acad Sci USA. 1987;84:4398–4402. doi: 10.1073/pnas.84.13.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz A W, Visscher J, Bakker C G, Niessen J. Origins Life. 1987;17:351–357. doi: 10.1007/BF02386473. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz A W, de Graaf R M. J Mol Evol. 1993;36:101–106. [Google Scholar]
- 9.Larralde R, Robertson M P, Miller S L. Proc Natl Acad Sci USA. 1995;92:8158–8160. doi: 10.1073/pnas.92.18.8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spach G. Origins Life. 1984;14:433–437. doi: 10.1007/BF00933688. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz A W, Orgel L E. Science. 1985;228:185–187. doi: 10.1126/science.228.4699.585. [DOI] [PubMed] [Google Scholar]
- 12.Orgel L E. Origins Life. 1986;17:27. doi: 10.1007/BF01809810. [DOI] [PubMed] [Google Scholar]
- 13.Cherny D Y, Belotserkovskii B P, Frank-Kamenetskii M D, Egholm M, Buchardt O, Berg R H, Nielsen P E. Proc Natl Acad Sci USA. 1993;90:1667–1670. doi: 10.1073/pnas.90.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen P E. Origins Life Evol Biosphere. 1993;23:327. doi: 10.1007/BF01582083. [DOI] [PubMed] [Google Scholar]
- 15.Pitsch S, Krishnamurthy R, Bolli M, Wendeborn S, Holzner A, Minton M, Lesueur C, Schlönvogt I, Juan B, Eschenmoser A. Helv Chim Acta. 1995;78:1621–1635. [Google Scholar]
- 16.Miller S L. Nat Struct Biol. 1995;4:167–169. doi: 10.1038/nsb0397-167. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro R. Origins Life Evol Biosphere. 1995;25:83–98. doi: 10.1007/BF01581575. [DOI] [PubMed] [Google Scholar]
- 18.Stoks P G, Schwartz A W. Geochim Cosmochim Acta. 1981;45:563–569. [Google Scholar]
- 19.Cronin J R, Pizzarello S, Cruikshank D P. In: Meteorites and the Early Solar System. Kerridge J S, Matthews M S, editors. Tucson, AZ: Univ. Arizona Press; 1988. pp. 819–857. [Google Scholar]
- 20.Stoks P G, Schwartz A W. Nature (London) 1979;282:709–710. [Google Scholar]
- 21.Miller S L. Cold Spring Harbor Symp Quant Biol. 1987;52:17–27. doi: 10.1101/sqb.1987.052.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Yuasa S, Flory D, Basile B, Oró J. J Mol Evol. 1984;21:76–80. doi: 10.1007/BF02100630. [DOI] [PubMed] [Google Scholar]
- 23.Ferris J P, Sanchez R A, Orgel L E. J Mol Biol. 1968;33:693–704. doi: 10.1016/0022-2836(68)90314-8. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez R A, Ferris J E, Orgel L E. Science. 1966;154:784–785. doi: 10.1126/science.154.3750.784. [DOI] [PubMed] [Google Scholar]
- 25.Kasting J F. Science. 1993;299:920–926. doi: 10.1126/science.11536547. [DOI] [PubMed] [Google Scholar]
- 26.Raulin, F. & Toupance, G. (1976) Bull. Soc. Chim. 29–39.
- 27.Miller S L, Orgel L E. The Origins of Life on the Earth. Englewood Cliffs, N. J.: Prentice Hall; 1974. [Google Scholar]
- 28.Hong J H, Pownall H J, Becker R S. Photochem Photobiol. 1976;24:217–222. [Google Scholar]
- 29.Sasaki, T., Yoshioka, T. & Shoji, K. (1969) J. Chem. Soc., 1086–1088.
- 30.Phibbs, M. K. & Sipos, P. A. (1980) Canada Patent 1,068,301; Chem. Abs.93, 45994p.
- 31.Truce W E, Tichenor G J W. J Org Chem. 1972;37:2391–2396. [Google Scholar]
- 32.Morita, K., Hashimoto, N. & Matsumura, K. (1975) Germany Patent 2,506,029; Chem. Abs.83, 194012n.
- 33.Ferris J P, Goldstein G, Beaulieu D L. J Am Chem Soc. 1970;92:6598–6603. [Google Scholar]
- 34.Ferris J P, Zamek O S, Altbuch A M, Freeman H J. J Mol Evol. 1974;3:301–309. doi: 10.1007/BF01796045. [DOI] [PubMed] [Google Scholar]
- 35.Sagan C, Thompson W R, Khare B N. Acc Chem Res. 1992;25:286–292. doi: 10.1021/ar00019a003. [DOI] [PubMed] [Google Scholar]
- 36.Irvine W M. Origins Life Evol Biosphere. 1998;28:365–383. doi: 10.1023/a:1006574110907. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y L, Lee H D, Beach M W, Margerum D W. Inorg Chem. 1987;26:2444–2449. [Google Scholar]
- 38.Kemp, I. A. & Kohnstam, G. J. (1956) J. Chem. Soc. 900–911.
- 39.Hirose Y, Ohmuro K, Saigoh M, Nakayama T, Yamagata Y. Origins Life Evol Biosphere. 1990–1991;20:471–481. [Google Scholar]
- 40.Robertson M P, Miller S L. Nature (London) 1995;375:772–774. doi: 10.1038/375772a0. , and correction, 377, 257. [DOI] [PubMed] [Google Scholar]
- 41.Browne, M. (1995) The New York Times, July 4, 1995, pp. C1 & C16.
- 42.Cieplak A S, Wiberg K B. J Am Chem Soc. 1992;114:9226–9227. [Google Scholar]
- 43.Raulin F, Bloch S, Toupance G. Origins Life. 1977;8:247–257. doi: 10.1007/BF00930686. [DOI] [PubMed] [Google Scholar]
- 44.Jacobson A R, Coffin S H, Shearson C M, Sayre L M. Mol Toxicol. 1987;1:17–34. [PubMed] [Google Scholar]
- 45.Sakurai S, Yanagawa H. Origins Life. 1984;14:171–176. doi: 10.1007/BF00933655. [DOI] [PubMed] [Google Scholar]
- 46.Nagayama Y, Takaoka O, Inomata K, Yamagata Y. Origins Life Evol Biosphere. 1990;20:249–257. doi: 10.1007/BF01808107. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro R, Klein R S. Biochemistry. 1966;5:2358–2362. doi: 10.1021/bi00871a026. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro R, Klein R S. Biochemistry. 1967;6:3576–3582. doi: 10.1021/bi00863a032. [DOI] [PubMed] [Google Scholar]
- 49.Lindahl T, Nyborg B. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 50.Lindahl T. Science. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 51.Duncan B K, Miller J H. Nature (London) 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 52.Garrett E R, Tsau J. J Pharm Sci. 1972;61:1052–1061. doi: 10.1002/jps.2600610703. [DOI] [PubMed] [Google Scholar]
- 53.Shapiro R, Servis R E, Welcher M. J Am Chem Soc. 1970;92:422–424. [Google Scholar]
- 54.Hayatsu H, Wataya Y, Kazushige K. J Am Chem Soc. 1970;92:724–726. doi: 10.1021/ja00706a062. [DOI] [PubMed] [Google Scholar]
- 55.Levy M, Miller S L. Proc Natl Acad Sci USA. 1998;95:7933–7938. doi: 10.1073/pnas.95.14.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shapiro R. In: Chromosome Damage and Repair. Seeberg E, Kleppe K, editors. New York: Plenum; 1981. pp. 3–18. [Google Scholar]
- 57.Frick L, Mac Neela J P, Wolfenden R. Bioorg Chem. 1987;15:100–108. [Google Scholar]
- 58.Frederico L A, Kunkel T A, Shaw B R. Biochemistry. 1990;29:2532–2537. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- 59.Blackburne G M, Gait M J. Nucleic Acids in Chermistry and Biology. 2nd Ed. Oxford: Oxford Univ. Press; 1996. pp. 310–312. [Google Scholar]
- 60.Stribling R, Miller S L. Origins Life. 1987;17:261–273. doi: 10.1007/BF02386466. [DOI] [PubMed] [Google Scholar]
- 61.Summers D P, Chang S. Nature (London) 1993;365:630–633. doi: 10.1038/365630a0. [DOI] [PubMed] [Google Scholar]
- 62.Lowe D R. In: Early Life on Earth. Bengtson S, editor. New York: Columbia Univ. Press; 1994. pp. 24–35. [Google Scholar]
- 63.Jones F E. Evaporation of Water, with Emphasis on Applications and Measurements. Chelsea, Michigan: Lewis Publishers; 1992. [Google Scholar]
- 64.Barnes R S K. Coastal Lagoons. Cambridge, U.K.: Cambridge Univ. Press; 1980. [Google Scholar]
- 65.Logan B W. The MacLeod Evaporite Basin, Western Australia. Tulsa, OK: Am. Assn. Petroleum Geologists; 1987. [Google Scholar]
- 66.Holland H D. The Chemical Evolution of the Atmosphere and Oceans. Princeton: Princeton Univ. Press; 1984. [Google Scholar]
- 67.Schreiber B C. In: in Evaporites and Hydrocarbons. Schreiber B C, editor. New York: Columbia Univ. Press; 1988. pp. 1–10. [Google Scholar]
- 68.Holm N G. Origins Life Evol Biosphere. 1992;22:5–14. [Google Scholar]
- 69.Clark B C. Origins Life Evol Biosphere. 1988;18:209–238. doi: 10.1007/BF01804671. [DOI] [PubMed] [Google Scholar]
- 70.Woese C. In: The Origins of Life and Evolution. Halvorson H O, Van Holde K E, editors. New York: Liss; 1980. pp. 65–76. [Google Scholar]
- 71.Shapiro R, Cohen B I, Servis R E. Nature (London) 1970;277:1047–1048. doi: 10.1038/2271047a0. [DOI] [PubMed] [Google Scholar]
- 72.Arrhenius G, Sales B, Mojzsis S, Lee T. J Theor Biol. 1997;187:503–522. doi: 10.1006/jtbi.1996.0385. [DOI] [PubMed] [Google Scholar]
- 73.Monod J. Le Hazard et la Necessité. Paris: Editions de seuil; 1970. ; trans. Wainhouse, A. G. (1971) Change and Necessity (Knopf, New York), pp. 144–145. [Google Scholar]
- 74.Cairns-Smith A G. Genetic Takeover and the Mineral Origins of Life. New York: Cambridge Univ. Press; 1982. [Google Scholar]
- 75.Dyson F. Origins of Life. Cambridge: Cambridge Univ. Press; 1985. [Google Scholar]
- 76.Kauffman S. The Origins of Order. Self-Organization and Selection in Evolution. New York: Oxford Univ. Press; 1993. [Google Scholar]
- 77.Wächtershäuser G. Prog Biophys Mol Biol. 1992;58:85–201. doi: 10.1016/0079-6107(92)90022-x. [DOI] [PubMed] [Google Scholar]