

Abstract
Genetic modification of cells and animals is an invaluable tool for biotechnology and biomedicine. Currently, integrating vectors are used for this purpose. These vectors, however, may lead to insertional mutagenesis and variable transgene expression and can undergo silencing. Scaffold/matrix attachment region-based vectors are nonviral expression systems that replicate autonomously in mammalian cells, thereby making possible safe and reliable genetic modification of higher eukaryotic cells and organisms. In this study, genetically modified pig fetuses were produced with the scaffold/matrix attachment region-based vector pEPI, delivered to embryos by the sperm-mediated gene transfer method. The pEPI vector was detected in 12 of 18 fetuses in the different tissues analyzed and was shown to be retained as an episome. The reporter gene encoded by the pEPI vector was expressed in 9 of 12 genetically modified fetuses. In positive animals, all tissues analyzed expressed the reporter gene; moreover in these tissues, the positive cells were on the average 79%. The high percentage of EGFP-expressing cells and the absence of mosaicism have important implications for biotechnological and biomedical applications. These results are an important step forward in animal transgenesis and can provide the basis for the future development of germ-line gene therapy.
Keywords: farm animals, gene therapy, sperm-mediated gene transfer, transgenesis
Genetically modified animals are invaluable tools as model systems in basic research and in applied fields such as gene therapy, xenotransplantation, bioreactors for the production of pharmaceutical or other commercial products, and the genetic improvement of livestock. Numerous vector systems are currently in use for the genetic modification of animals but they all rely on the integration of foreign genes into the animal genome. Ideally, a vector compatible with all applications should fulfill at least the following requirements: (i) it should be stably retained, both structurally and mitotically, in the genetically modified animal; (ii) it should not contain viral DNA sequences or encode for viral proteins that bear the risk of cellular transformation; and (iii) there should be long-term expression of the vector-encoded transgene (1, 2). In addition, the ideal vector should be highly effective in the production of genetically modified animals and transmission of the transgene to the offspring, although the latter is not a requirement necessary for all applications. These requirements are not trivial to achieve with vectors currently used in gene therapy and animal transgenic technology (3) such as plasmid-based DNA vectors, viral integrating vectors, and episomally replicating viral vectors, namely simian virus 40- or EBV-derived vectors (4–8). Therefore, there is an increasing consensus that, as an alternative approach, nonintegrating, nonviral vector systems could fulfill most of the requirements for safe and reproducible modification of eukaryotic cells and organisms (2, 6, 9, 10). The development of such vectors has encountered serious problems: naturally occurring mammalian plasmids have never been observed and cloning of putative mammalian origins of replications (ORIs), akin to prokaryotic ORIs or yeast autonomously replicating sequences, has not yielded unequivocal and reproducible results (reviewed in refs. 6 and 9). In recent years, efforts have been made to develop mammalian artificial chromosomes of megabase size that replicate autonomously alongside the host chromosomes and are mitotically stable in the absence of selection (10). Such vectors can accommodate large inserts and are free of the problems caused by viral vectors, although some artificial chromosomes have been constructed and used as models for gene therapy and the production of transgenic animals expressing complex proteins (refs. 11–14 and for review refs. 9 and 10). In this approach the technical difficulties connected with preparing, manipulating, and delivering such large molecules will have to be overcome.
A much more promising nonviral episomal vector system has recently been developed that bypasses the requirement for a canonical mammalian origin of replications (15, 16). In the pEPI vector family both replication and mitotic stability of the plasmid are assured by the presence of a human scaffold/matrix attachment region (S/MAR) linked to a transcription unit (16). Such vectors have been shown to replicate in a variety of cell lines and primary cells at a copy number of 5–10 per cell and to be stably maintained in the absence of selection for over hundreds of generations (15–18); furthermore, vector transgenes encoded by these vectors are not subject to epigenetic silencing (18, 19). For these reasons, this vector system would appear to be ideal for gene therapy and the generation of genetically modified animals. To achieve these goals pEPI vectors must be capable of replicating and expressing gene products in differentiating cells and developing and growing animal tissues. This proposition is the object of the present study.
As a test animal we chose the pig, in view of its being representative of the large farm animals that are used in most applications of animal transgenesis, and in particular as preclinical models of human diseases. Several methods are currently available for generating transgenic animals (20, 21). The sperm-mediated gene transfer (SMGT) method, originally developed in mouse (22), has been shown to function consistently with high efficiency in many animal species including swine (23–25), in which other methods function at low efficiency (26, 27). In this article we show that pEPI can be delivered with high efficiency to the pig embryo by SMGT, that the transgene is expressed in all tissues of positive fetuses that were tested, and that it is retained as an episome in the course of embryogenesis and fetal development. We have successfully attempted to produce a genetically modified animal by using a nonviral episomal vector. Another important consideration is that the results of this study can provide the basis for the future development of germ-line gene therapy.
Results
Sperm-DNA Uptake and Production of Genetically Modified Pigs Using pEPI-EGFP.
A Large White boar was used as sperm donor and DNA uptake was assessed, as described (28), by scintillation counting in time-course experiments using pEPI-EGFP (Fig. 1 A) and its progenitor commercial plasmid pGFP-C1 (Fig. 4, which is published as supporting information on the PNAS web site). The uptake kinetics of the pEPI vector was identical to those of pGFP-C1 and was very similar to those described for other plasmids (28); there was rapid binding of most of the DNA during the initial 15–30 min, followed 60 min later by a plateau. The most appropriate DNA incubation conditions for sperm cells were determined as described (28). Sperm cells were used as vectors for transferring pEPI-EGFP into eggs by laparoscopic insemination. Eighteen fetuses were harvested from two sows on day 70 of pregnancy. This time period corresponds to over 2/3 of pregnancy, when cell differentiation and organogenesis are fully completed after which only growth of the organism takes place. An additional nine fetuses were produced by a fertilization performed with sperm that had not been incubated with exogenous DNA (negative controls). Tissue samples were recovered from skeletal muscle, heart, liver, kidney, and lung of fetuses obtained from all three fertilizations and studied for the presence and expression of the transgene.
Fig. 1.

Map of pEPI-EGFP and examples of PCR analysis. (A) Map of pEPI-EGFP. pEPI-EGFP derives from the commercial plasmid pGFP-C1 (Clontech). An S/MAR sequence, obtained from the human IFN β-gene, is contained in the multiple cloning site (MCS). The Neo/Kan gene is driven by dual promoters to confer kanamycin resistance in bacteria and G418 resistance in mammalian cells. EGFP is the enhanced version of the GFP gene. BglII and EcoRI restriction sites are indicated. SV40, simian virus 40; ori, origin of replication; HSV, herpes simplex virus. (B) Examples of PCR analysis. PCR was performed on total DNA extracted from liver tissues after separation of nuclei with EGFP-specific primers. Lanes 2–4 indicate three samples that tested positive (fetuses 1, 2, and 8). Hirt extracts were also analyzed by PCR. Lanes 5–7 indicate fetuses that tested positive (fetuses 9, 10, and 11). Lane 1 is a negative control (WT fetus). M, 100-bp DNA Ladder (New England Biolabs, Milan, Italy); P, positive control, pEPI-EGFP amplification.
pEPI-EGFP Is Maintained in the Episomal State.
To determine whether the nonviral vector used to genetically modify pigs by SMGT was retained in the fetuses, total DNA was prepared from skeletal muscle, heart, liver, kidney, and lung tissues. Three to five tissues from the fetuses were analyzed by PCR (43 tissues in total) using primers specific for EGFP, pCMV, and Neo/Kan regions of the plasmid. The presence of pEPI-EGFP could be demonstrated in 12 of 18 fetuses (67%). In these 12 fetuses the presence of the vector could be demonstrated in all tissues analyzed (Table 1 and Table 2, which is published as supporting information on the PNAS web site; examples of EGFP amplifications are shown in Fig. 1B, lanes 2–4). A first indication of whether pEPI-EGFP was retained as an episome came from PCR analysis of Hirt DNA extracts that selectively contain extrachromosomal DNA. Such analysis, performed on fetal tissues that had been shown to contain pEPI-EGFP vector DNA, revealed the presence of pEPI-EGFP in the Hirt extracts from all 37 tissues examined (Fig. 1 B, lanes 5–7 and Table 1; for details see Table 2). To further demonstrate that pEPI-EGFP was retained as an episome during fetal development, Southern blot analysis using total DNA or DNA from the Hirt extracts prepared from muscle, liver, heart, and kidney was performed on four fetuses that had tested positive in the PCR analyses. Either undigested DNA or DNA digested with BglII, which linearizes pEPI-EGFP, was loaded onto agarose gels and hybridized with pEPI-EGFP as a probe. The DNA band pattern was identical in all cases: in undigested DNA two bands were found, corresponding to the open circle and supercoiled forms of pEPI-EGFP, whereas in BglII-digested DNA one band could be seen, corresponding to the linearized form of the vector (Fig. 2 A). The intensity of these bands was weak, and by comparing it to the hybridization signal of isolated vector DNA at different concentrations, the vector copy number was estimated to be <10 per cell, a number consistent with that found in cell lines transfected with the pEPI vector (15–18). In no case was hybridization to high molecular weight DNA observed, strongly suggesting that no integration events had occurred. The episomal state was further confirmed by rescue experiments in which Escherichia coli was transformed with DNA isolated from Hirt extracts prepared from two tissues of four fetuses. Kanamycin-resistant bacterial colonies were obtained from samples of heart and skeletal muscle (fetus 2), liver and skeletal muscle (fetuses 6 and 9), and liver and kidney (fetus 10), although, because of the low copy number of the vector, only very few (between 3 and 10) colonies were obtained in these rescue experiments. Plasmid DNA extracted from these clones was digested with BglII (Fig. 2 B, lane 3) or double digested with BglII and EcoRI (Fig. 2 B, lane 4) and analyzed on agarose gels. In all cases the restriction patterns were identical to those observed in digested pEPI-EGFP plasmid vector (Fig. 2 B, lanes 1 and 2).
Table 1.
Efficiency of SMGT method to produce genetically modified pig fetuses by using the nonviral episomal plasmid pEPI-EGFP
| Fetus | Tissues analyzed,no. | DNA |
RNA | Protein | |
|---|---|---|---|---|---|
| Total | Extrachromosomal | ||||
| M | 1 | − | − | − | − |
| C | 1 | − | − | − | − |
| 1 | 3 | + | + | + | + |
| 2 | 5 | + | + | + | + |
| 3 | 4 | + | + | + | + |
| 4 | 3 | − | − | − | − |
| 5 | 5 | − | − | − | − |
| 6 | 5 | + | + | + | + |
| 7 | 3 | − | − | − | − |
| 8 | 4 | + | + | + | + |
| 9 | 4 | + | + | + | + |
| 10 | 4 | + | + | + | + |
| 11 | 4 | + | + | + | + |
| 12 | 3 | + | + | + | + |
| 13 | 2 | + | + | − | − |
| 14 | 2 | + | + | − | − |
| 15 | 3 | + | + | − | − |
| 16 | 2 | − | − | − | − |
| 17 | 2 | − | − | − | − |
| 18 | 2 | − | − | − | − |
| Total, % | 62 | 12/18 (67) | 12/18 (67) | 9/12 (75) | 9/12 (75) |
+ and − indicate the presence or absence of EGFP sequence or gene product. Total DNA extracted from nuclei prepared from different tissues was analyzed by PCR using three set of primers specific for EGFP, pCMV, and Neo/Kan sequences. Extrachromosomal DNA extracted by the Hirt method was also analyzed by PCR, Southern blotting, and plasmid rescue. RNA and proteins extracted from tissue biopsies were subjected to RT-PCR and Western blotting, respectively. Tissue sections were subjected to confocal microscopy. C, tissue samples from negative control fetus. M, tissue samples from the mother of one offspring, as negative control.
Fig. 2.

pEPI-EGFP is episomal in transgenic tissues. (A) Southern blot analysis. M, 1-Kbp Ladder (O'Gene Ruler; Fermentas, St. Leon-Rot, Germany); P, positive control (250 pg of linearized pEPI-EGFP vector DNA). BglII-linearized Hirt extracts from fetus 6 (liver, lane 1), fetus 9 (muscle, lane 3), and fetus 10 (kidney, lane 5; liver, lane 7) or undigested Hirt DNA from the same fetuses (lanes 2, 4, 6, and 8, respectively) are shown. (B) Example of restriction analysis of rescued plasmid (fetus 6, liver). M, 1-Kbp Ladder (O'Gene Ruler, Fermentas). Lane 1, control pEPI-EGFP linearized with BglII; lane 2, control pEPI-EGFP digested with BglII and EcoRI that removes the S/MAR; lane 3, rescued linearized pEPI-EGFP (BglII); lane 4, rescued double-digested plasmid (BglII and EcoRI).
pEPI-EGFP Is Expressed in Tissues of Genetically Modified Pigs
Expression of the EGFP reporter gene was assessed by RT-PCR analysis. Total RNA was prepared from skeletal muscle, heart, liver, kidney, and lung of the 12 fetuses that had been shown to be genetically modified and assayed for the presence of the EGFP transcript. In nine fetuses (75%) the EGFP transcript was detected in all 36 tissues analyzed (three to five tissues per fetus were tested; Tables 1 and 2). Fig. 3A provides examples of such an RT-PCR analysis using RNA from liver, skeletal muscle, and heart of genetically modified fetuses and two control fetuses. The primers used, designed on the EGFP gene, amplified the expected 480-bp fragment. In no case was an RT-PCR product amplified by using RNA from tissues of control fetuses (Fig. 3 A). GAPDH amplification was performed to control the RNA preparations.
Fig. 3.

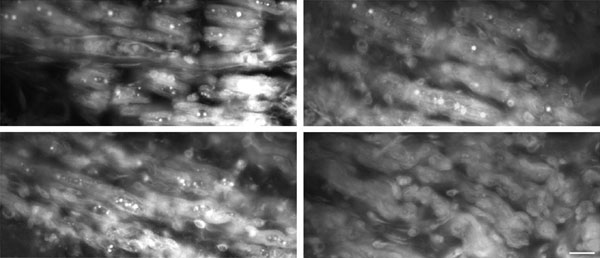
Analysis of EGFP expression in genetically modified fetuses. (A) RT-PCR. (Upper) M, 100-bp DNA Ladder (New England Biolabs); C, negative control. Lanes 1–5, fetuses 1, 2, 3, 6, and 8, respectively; lane 6, negative fetus 5. In every case liver was the tested organ. (Lower) M, 100-bp DNA Ladder (New England Biolabs; C, negative control; lanes 1–4 are fetuses 9, 10, 11, and 12; lane 5, negative fetus 13. Lanes C and 1, 2, and 5 are muscle samples; lanes 3 and 4 are heart samples. EGFP expression was compared with expression of the endogenous gene GAPDH. (B) Western blot analysis of protein extracts from a representative genetically modified fetus. EGFP protein expression from tissues of genetically modified fetus 6 and a control fetus was analyzed on 12% SDS/PAGE, blotted, and probed with anti-EGFP polyclonal antibody that recognizes a specific 26-kDa band (Upper) and with an anti-β-actin mAb that recognizes a specific 42-kDa band (Lower). One hundred fifty micrograms of protein extracts was loaded on lanes 1–6. Lane 1, WT fetus, liver; lanes 2–6, fetus 6, skeletal muscle, heart, liver, kidney, and lung. Seventy-five micrograms of protein extracts from COS7 cells transfected with pGFP-C1 was used as positive control (lane C). (C) Confocal microscopy. Micrograph of six tissues (from left to right: skeletal muscle, heart, liver, kidney, lung, and skin) from the representative positive fetus 6 (Upper) and the same tissues from the negative fetus 5 (Lower). (Magnification: ×20; bar: 20 microns.)
EGFP protein expression was assessed by Western blot analysis on skeletal muscle, heart, liver, kidney, and lung protein extracts. Protein extracts were fractionated on SDS/PAGE, blotted onto nitrocellulose membranes, and probed with polyclonal anti-EGFP antibody that recognizes a 26-kDa band. EGFP expression was demonstrated in all 34 tissues analyzed (three to five per fetus) of the nine fetuses that had tested positive for the presence of the EGFP transcript (Table 1 and 2). Fig. 3B shows the Western blot analysis of protein extracts from the five tissues of a representative fetus. The presence of the EGFP protein was also analyzed by epifluorescence and confocal microscopy. Fig. 3C shows confocal micrographs of the five tissues from a representative positive (Upper) and negative fetus (Lower). EGFP expression in the skin is shown in Fig. 3C and all embryonic layer-derived tissues are represented.
The tissues that had tested positive in Western blot analysis also tested positive by microscopy (Table 2). The percentage of positive cells in the various tissues is presented in Table 3, which is published as supporting information on the PNAS web site. EGFP expression was observed in 62–88% of the cells from the different samples (78.8 ± 5.4%, mean ± SD). There were no significant differences in these percentages between the different fetuses and the different combined tissues (one-way ANOVA P = 0.929 and P= 0.091, respectively). Protein expression was seen as a diffuse pattern in all samples analyzed; at high magnification muscle fibers also presented discrete intense fluorescent spots (Fig. 5, which is published as supporting information on the PNAS web site). Fluorescence emission revealed no expression in control tissues. Histological analysis of all tissues from the different animals displayed a normal appearance (data not shown).
Discussion
The aim of this study was to test whether a nonviral episomal vector could be used to generate a genetically modified animal. Such an episomal vector could overcome most of the problems connected with the use of integrating vectors currently used for the genetic modification of animals. The method chosen to genetically modify the animals, SMGT, has been shown to produce transgenic animals with high efficiency (24, 28). This approach to transgenesis offers other advantages for the production of genetically modified animals: it does not require embryo handling and can be performed with simple techniques and at low costs.
This study demonstrates that the episomal pEPI-EGFP vector can be taken up by sperm cells and subsequently introduced into the oocyte at fertilization, generating genetically modified pigs. To date, SMGT experiments have been carried out mostly with enzymatically linearized plasmid DNA that became integrated in the sperm genome and that was subsequently introduced as part of the sperm genome into the oocyte. When nonepisomal covalently closed plasmid was incubated with sperm, the DNA was taken up by the spermatozoa, was integrated in the genome with reduced efficiency, and was frequently subject to sequence rearrangements (ref. 29 and M.L., unpublished results). In the present study the pEPI-EGFP episomal plasmid was used in its covalent form, which resulted in the production of genetically modified pigs with an efficiency as high as that previously obtained with linearized plasmid (24, 28).
We concentrated our analyses on skeletal muscle, heart, liver, kidney, and lung because these tissues are expected to be the main targets for therapeutic approaches. We have also studied transgene expression at the protein level in skin so as to have expression data on tissues derived from all three embryonic layers. pEPI-EGFP DNA was found in 43 tissues of 12 of the 18 fetuses generated, and its episomal status was demonstrated by PCR, Southern blot, and plasmid rescue analyses of Hirt extracts. In Southern blot analyses the pEPI-EGFP vector could be detected in its circular form and, when linearized, exhibited the same size as the input DNA. In these analyses no integration of the vector into the host genome could be observed. This finding strongly suggests that if some such events do occur they are rare and must take place during the final stages of embryogenesis. Moreover, analysis of the vector DNA recovered from the rescue experiments demonstrated that no rearrangements of the vector had occurred during cell differentiation. The fact that pEPI was shown to be episomal in all of the tissues analyzed has important implications for applications in gene and cell therapy.
The episomal state of the plasmid in the fetuses generated by SMGT shows that the presence of S/MAR DNA sequences, which ensures episomality of the plasmid in cultured cells, also prevents integration of the plasmid into the genome of sperm cells and subsequently in the cells of the individuals generated by that sperm. An interesting conclusion provided by this result is that integration of the vector into the sperm genome is not required for proper functioning of SMGT, nor is it responsible for the high efficiency of the method. In the specific case of pEPI, association of the vector with the sperm chromatin throughout the different stages of pronuclear development and formation of the zygotic nucleus is most likely assured by the S/MAR sequences (30, 31). In 75% of the genetically modified fetuses the pEPI vector also expressed the EGFP gene, as attested by RT-PCR, Western blotting, and confocal microscopy.
Three of the 12 fetuses positive for the presence of pEPI-EGFP DNA did not show detectable transgene expression at the RNA or protein levels (Tables 1 and 2). A possible explanation for this observation is that the vector may be present at a very low copy number in the cells: in these fetuses transcription could occur at a basal level sufficient for vector maintenance (32) but insufficient for the detection of gene products.
Because the proportion of positive cells in the various tissues is a central issue for the fidelity of passage of the pEPI plasmid from the original zygote down through many cell divisions into the various embryonic layers and then the fetus, we carried out a detailed analysis of the tissues. In positive animals, all tissues analyzed expressed EGFP; moreover in these tissues, the positive cells were 78.8% on the average. The high percentage of EGFP-expressing cells and the absence of mosaicism have important implications for biotechnological and biomedical applications.
In this study we did not investigate the germ-line transmission of pEPI vector, and at this stage we cannot rule out the possibility that this vector type will not be properly transmitted to the next generation. Although we observed by RT-PCR, Western blot, and microscopy analyses efficient expression of the reporter gene EGFP in all tissues tested, expression efficiencies of other target genes still have to be determined. Nevertheless, the results reported here are an important step forward in the genetic modification of animals. Moreover, the successful transfer of an episomal vector by SMGT to embryos, followed by the development of normal fetuses containing the vector in all tissues in the episomal state, can provide the basis for the future development of germ-line gene therapy, which would allow the treatment of genetic diseases at the time of conception. The episomal nature of the vector would overcome the problems of safety that have plagued efforts to apply gene therapy to humans by using integrating vectors and would ensure the distribution of the vector to most, if not all, districts of the organism.
Methods
Plasmid Vectors.
The plasmid used in this study was pEPI-EGFP (Fig. 1 A), derived from the commercial plasmid pGFP-C1 (Clontech, Mountain View, CA), which was used in some control experiments. pEPI-EGFP and pGFP-C1 are identical except that pEPI-EGFP contains a 2-Kbp S/MAR sequence, obtained from the human IFN β-gene, in the polylinker site and that the GFP gene has been replaced in the same location by its enhanced version, EGFP. The plasmids were amplified in E. coli NovaBlue (Novagen/Calbiochem, San Diego, CA).
Animals.
Semen was collected from a trained Large White boar that had abstained for 3 days. Large White recipient prepubertal gilts (99 ± 1.80 kg) were superovulated, synchronized, and surgical laparoscopic-inseminated at the utero-tubal junction (1 × 10 9 DNA treated sperm per gilt) 36 h after human chorionic gonadotropin injection as described (33). Animal care and experimental procedures met local, national, and European Union Guidelines.
Preparation of Sperm and DNA Uptake.
Semen was collected and prepared as reported (24, 28) with minor modifications. Briefly, immediately after collection, semen was diluted 1:1 with swine fertilization medium (SFM) [11.25 g of glucose, 10 g of sodium citrate (2H2), 4.7 g of EDTA (2H2O), 3.25 g of citric acid (H2O), and 6.5 g of Trizma per liter] adjusted in this experiment to pH 6.8, prewarmed to 37°C. Seminal fluid was removed by rediluting the sperm suspension 1:10 with SFM and centrifuging it (800 × g for 10 min) in 50-ml Falcon tubes (Becton Dickinson, Milan, Italy). The washing procedure was repeated once again with SFM supplemented with 6 mg/ml BSA (Fraction V; Sigma-Aldrich, Milan, Italy) (SFM/BSA) prewarmed to 25°C. Sperm cells were counted with a hemocytometric chamber and resuspended at a working dilution of 1 × 108 cells per ml in 25°C SFM/BSA.
To determine the most appropriate sperm-DNA incubation conditions, parallel time-course experiments were performed at different temperatures (17°C, 20°C, 25°C, or 37°C) or with increasing amounts of DNA, and DNA uptake was assessed by scintillation counting, as described (28). Washed ejaculated sperm cells, resuspended at a concentration of 1 × 108 cells per ml of SFM/BSA, were mixed with pEPI-EGFP or pGFP-C1 DNA labeled by nick translation (34). Aliquots containing 1 × 106 sperm cells were withdrawn from the incubation mixture at specific times, diluted in Eppendorf (Hamburg, Germany) tubes containing 1 ml of SFM and washed twice by centrifuging at 3,000 × g for 5 min.
Production of Genetically Modified Pigs.
Washed sperm cells were incubated for 1 h at 17°C with circular pEPI-EGFP plasmid in SFM/BSA (5 μg of DNA per 108 spermatozoa per ml). Tubes were inverted every 20 min to prevent sperm sedimentation. The final 20 min of incubation was at room temperature followed by heating (37°C) for 1 min just before surgery. Laparoscopic insemination (33) was performed with 5-ml aliquots per uterine horn, containing 5 × 108 DNA-treated spermatozoa. Surgical harvest of fetuses was performed under total anesthesia on day 70 of pregnancy.
Preparation of DNA and RNA.
DNA and RNA were prepared from frozen tissues (skeletal muscle, heart, liver, kidney, and lung). Total DNA was prepared from tissue sections after separation of nuclei, following standard protocols (34). Hirt extraction of extrachromosomal DNA from tissue sections was also performed as described (35). DNA was precipitated overnight at −20°C after addition of 0.1 volume of 3 M sodium acetate and 2.5 volumes of ethanol. DNA was recovered by centrifugation and resuspended in 0.1× TE (1 mM EDTA/10 mM Tris·HCl, pH 7.5). Total RNA was extracted by using the Versagene RNA Tissue kit according to the manufacturer's protocol (Gentra, Milan, Italy).
PCR, RT-PCR, and Southern Blot Analysis.
Both total DNA and Hirt-extracted DNA were analyzed by PCR. All of the primers used in this study were designed with Primer3 software (36). Eighty nanograms of DNA was amplified with: EGFP primers (Sigma-Genosys): 5′-CCT GAA GTT CAT CTG CAC CA-3′ (forward), 5′-TGC TCA GGT AGT GGT TGT CG-3′ (reverse); pCMV primers (Sigma-Genosys): 5′-CGT CAA TGG GTG GAG TAT TT-3′ (forward), 5′-AAT GGG GTG GAG ACT TGG AA-3′ (reverse); and Neo/Kan primers (Sigma-Genosys) 5′-GGC TAT TCG GCT ATG ACT GG-3′ (forward), 5′-GGA TAC TTT CTC GGC AGG AG-3′ (reverse). PCR was driven by AmpliTaq Gold (Applied Biosystems, Milan, Italy). Of the total RNA, 2 μg was reverse-transcribed with a SuperScript III kit (Invitrogen, Milan, Italy). The cDNA obtained was amplified by using the EGFP primers and primers specific for GAPDH porcine endogenous sequence: 5′-CAT CTT CCA GGA GCG AGA TCC C-3′ (forward), 5′-GTC AGG TCC ACA ACC GAC ACG-3′ (reverse). PCR products were analyzed on 1.5% TAE (Tris-acetate-EDTA)-agarose gels stained with ethidium bromide. The primers amplified a 480-bp fragment for EGFP and a 512-bp fragment for GAPDH. PCR and RT-PCR experiments were conducted in triplicate and subjected to routine controls. The risk of contaminating genomic DNA coamplification was ruled out by running the PCRs without prior reverse transcription. Total DNA and Hirt-extracted DNA were analyzed by Southern blot. Twenty micrograms of DNA, undigested, BglII-digested, or double-digested with BglII and EcoR, were fractionated on 0.7% agarose gels and blotted onto nylon membranes (32, 37). pEPI-EGFP vector was labeled with 32P (Ready-to-Go labeling kit, Amersham Pharmacia, Munich, Germany) and used as a probe. Hybridization was carried out in Church buffer (0.25 M sodium phosphate buffer, pH 7.2/1 mM EDTA/1% BSA/7% SDS) at 65°C for 16 h. Under our stringency conditions, we observed no hybridization in DNA samples from control fetuses.
Rescue Experiments.
Transformation of E. coli with DNA prepared by Hirt extraction was performed as described (15). Transformed colonies were selected on agarose plates containing 30 μg/ml kanamycin. DNA was isolated from individual resistant clones, subjected to restriction analysis, and subsequently fractionated and visualized on 1.5% agarose gels.
Western Blot.
Frozen tissues (skeletal muscle, heart, liver, kidney, and lung) were sonicated in lysis buffer (50 mM Hepes/10% glycerol/10 mM NaCl/10 mM DTT/1% SDS/5 mM EDTA) supplemented with 2% Protease Inhibitor Mixture (Sigma-Aldrich). Tissue debris were removed by centrifugation at 10,000 × g for 5 min. Resulting protein extracts were quantified by using the Bradford method (Bio-Rad, Milan, Italy) according to manufacturer's protocol, after which 150 μg of each protein extract was resolved in a 12% SDS/PAGE gel. After electroblotting to a nitrocellulose membrane (Amersham Biosciences, Milan, Italy), the membrane was probed with a polyclonal anti-EGFP antibody (MBL-Eppendorf, Milan, Italy), and signals were revealed by using a chemiluminescence system (ECL, Amersham Biosciences). To demonstrate equal loading of all lanes, membranes were probed with monoclonal anti-β-actin endogenous protein (AC15 clone; Sigma-Aldrich).
Microscopy.
Skeletal muscle, heart, liver, kidney, lung, and skin tissue samples were fixed with 4% paraformaldehyde (Sigma-Aldrich, Milan, Italy) in 0.01 M PBS, pH 7.4 at 4°C for 2 h, thoroughly washed in PBS at 4°C overnight, then embedded in OCT Matrix (CellPath; Hemel, Hempstead, UK) and quickly frozen in chilled isopentane in dry ice. Cryostat sections (30 μm) were cut and mounted on chrome-alum gelatin-coated slides, left to dry in a dust-free cooled cabinet, coverslipped, and sealed with Vectashield (Vector Labs, Burlingame, CA). Slides were initially analyzed with an Axioskope 2 epifluorescence microscope (Zeiss, Göttingen, Germany), equipped with a high-resolution digital camera (C4742–95, Hamamatsu Photonics, Milan, Italy), and HiPic software (Hamamatsu Photonics, Herrsching am Ammersee, Germany). For a detailed analysis a laser scanning microscope (LSM 510 Meta; Zeiss, Oberkochen, Germany) was used. Expression of the fluorescent protein was imaged with ×20, ×40, ×63, and ×100 oil immersion objectives. Images were captured at a resolution of 512 × 512 pixels. The appropriate argon laser fluorescence for visualization of the fluorophore, with an excitation wavelength of 488 nm and emission filter LP 560 was used. Images were adjusted for brightness and contrast and assembled as plates using Adobe PhotoShop (version 6.0; Adobe Systems, San Jose, CA). The number of positive cells per area was accomplished by a computer-assisted image analysis system (MCID 7.0; Imaging Res. Inc, St. Catharines, Canada) and expressed as a percentage. Six randomly selected sections of 200 × 200 μm each for each sample were analyzed.
Statistical Analysis.
All values are presented as means ± SD. Comparison of percentage of EGFP-positive cells (percentage of positive cells per area in six randomly selected sections) between the different fetuses and between the different combined tissues was performed with one-way ANOVA (SPSS 13.0; SPSS, Chicago, IL). Differences were considered significant at P < 0.05.
Supplementary Material
Acknowledgments
This work was supported by Italian Minister of Research and University Grant DD 21.09.99 n462 ric, University of Milano-Bicocca Grants Fondo Ateneo Ricerca 2003, 2004, and 2005 (to M.L.), and grants from the European Union and Deutsche Forschungsgemeinschaft (to H.J.L.).
Abbreviations
- SFM
swine fertilization medium
- S/MAR
scaffold/matrix attachment region
- SMGT
sperm-mediated gene transfer.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Lipps HJ, Bode J. Curr Opin Mol Ther. 2001;3:133–141. [PubMed] [Google Scholar]
- 2.Conese M, Auriche C, Ascenzioni F. Gene Ther. 2004;11:1735–1741. doi: 10.1038/sj.gt.3302362. [DOI] [PubMed] [Google Scholar]
- 3.Niemann H, Kues WA. Anim Reprod Sci. 2003;79:291–317. doi: 10.1016/s0378-4320(03)00169-6. [DOI] [PubMed] [Google Scholar]
- 4.Teschendorf C, Warrington KH, Jr, Siemann DW, Muzyczka N. Anticancer Res. 2002;22:3325–3330. [PubMed] [Google Scholar]
- 5.Kaiser J. Science. 2003;299:457–608. [Google Scholar]
- 6.Glover DJ, Lipps HJ, Jans DA. Nat Rev Genet. 2005;6:299–310. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 7.Ali SH, Kasper JS, Arai T, DeCaprio JA. J Virol. 2004;78:2749–2757. doi: 10.1128/JVI.78.6.2749-2757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humme S, Reisbach G, Feederle R, Delecluse HJ, Bousset K, Hammerschmidt W, Schepers A. Proc Natl Acad Sci USA. 2003;100:10989–10994. doi: 10.1073/pnas.1832776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipps HJ, Jenke AC, Nehlsen K, Scinteie MF, Stehle IM, Bode J. Gene. 2003;304:23–33. doi: 10.1016/s0378-1119(02)01215-5. [DOI] [PubMed] [Google Scholar]
- 10.Basu J, Willard HF. Trends Mol Med. 2005;11:251–258. doi: 10.1016/j.molmed.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Auriche C, Carpani D, Conese M, Caci E, Zegarra-Moran O, Donini P, Ascenzioni F. EMBO Rep. 2002;3:862–868. doi: 10.1093/embo-reports/kvf174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poggiali P, Scoarughi GL, Lavitrano M, Donini P, Cimmino C. Biochimie. 2002;84:1143–1150. doi: 10.1016/s0300-9084(02)00019-6. [DOI] [PubMed] [Google Scholar]
- 13.Robl JM, Kasinathan P, Sullivan E, Kuroiwa Y, Tomizuka K, Ishida I. Theriogenology. 2003;59:107–113. doi: 10.1016/s0093-691x(02)01262-1. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki N, Nishii K, Okazaki T, Ikeno M. J Biol Chem. 2006;281:26615–26623. doi: 10.1074/jbc.M603053200. [DOI] [PubMed] [Google Scholar]
- 15.Piechaczek C, Fetzer C, Baiker A, Bode J, Lipps HJ. Nucleic Acids Res. 1999;27:426–428. doi: 10.1093/nar/27.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenke AC, Stehle IM, Herrmann F, Eisenberger T, Baiker A, Bode J, Fackelmayer FO, Lipps HJ. Proc Natl Acad Sci USA. 2004;101:11322–11327. doi: 10.1073/pnas.0401355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaarschmidt D, Baltin J, Stehle IM, Lipps HJ, Knippers R. EMBO J. 2004;23:191–201. doi: 10.1038/sj.emboj.7600029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papapetrou EP, Ziros PG, Micheva ID, Zoumbos NC, Athanassiadou A. Gene Ther. 2006;13:40–51. doi: 10.1038/sj.gt.3302593. [DOI] [PubMed] [Google Scholar]
- 19.Jenke AC, Scinteie MF, Stehle IM, Lipps HJ. Mol Biol Rep. 2004;31:85–90. doi: 10.1023/b:mole.0000031363.35839.46. [DOI] [PubMed] [Google Scholar]
- 20.Clark J, Whitelaw B. Nat Rev Genet. 2003;4:825–833. doi: 10.1038/nrg1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter CV, Tiley LS, Sang HM. Trends Mol Med. 2005;11:293–298. doi: 10.1016/j.molmed.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Lavitrano M, Camaioni A, Fazio VM, Dolci S, Farace MG, Spadafora C. Cell. 1989;57:717–723. doi: 10.1016/0092-8674(89)90787-3. [DOI] [PubMed] [Google Scholar]
- 23.Lavitrano M, Lulli V, Maione B, Sperandio S, Spadafora C. Microinjection and Transgenesis: Strategies and Protocols. Springer, Heidelberg, Germany: Cid-Arregui A, García-Carrancá A; 1998. pp. 229–254. [Google Scholar]
- 24.Lavitrano M, Bacci ML, Forni M, Lazzereschi D, Di Stefano C, Fioretti D, Giancotti P, Marfe G, Pucci L, Renzi L, et al. Proc Natl Acad Sci USA. 2002;99:14230–14235. doi: 10.1073/pnas.222550299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavitrano M, Busnelli M, Cerrito MG, Giovannoni R, Manzini S, Vargiolu A. Reprod Fertil Dev. 2006;18:19–23. doi: 10.1071/rd05124. [DOI] [PubMed] [Google Scholar]
- 26.Wall RJ. Transgenic Res. 1999;8:313–315. doi: 10.1023/a:1008922129156. [DOI] [PubMed] [Google Scholar]
- 27.Wall RJ. Theriogenology. 2002;57:189–201. doi: 10.1016/s0093-691x(01)00666-5. [DOI] [PubMed] [Google Scholar]
- 28.Lavitrano M, Forni M, Bacci ML, Di Stefano C, Varzi V, Wang H, Seren E. Mol Reprod Dev. 2003;64:284–291. doi: 10.1002/mrd.10230. [DOI] [PubMed] [Google Scholar]
- 29.Maione B, Lavitrano M, Spadafora C, Kiessling AA. Mol Reprod Dev. 1998;50:406–409. doi: 10.1002/(SICI)1098-2795(199808)50:4<406::AID-MRD4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 30.Baiker A, Maercker C, Piechaczek C, Schmidt SB, Bode J, Benham C, Lipps HJ. Nat Cell Biol. 2000;2:182–1844. doi: 10.1038/35004061. [DOI] [PubMed] [Google Scholar]
- 31.Jenke BH, Fetzer CP, Stehle IM, Jonsson F, Fackelmayer FO, Conradt H, Bode J, Lipps HJ. EMBO Rep. 2002;3:349–354. doi: 10.1093/embo-reports/kvf070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stehle IM, Scinteie MF, Baiker A, Jenke AC, Lipps HJ. Chromosome Res. 2003;11:413–421. doi: 10.1023/a:1024962308071. [DOI] [PubMed] [Google Scholar]
- 33.Fantinati P, Zannoni A, Bernardini C, Webster N, Lavitrano M, Forni M, Seren E, Bacci ML. Theriogenology. 2005;63:806–817. doi: 10.1016/j.theriogenology.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2001. [Google Scholar]
- 35.Hirt B. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 36.Rozen S, Skaletsky HJ. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana, Totowa, NJ: Krawetz S, Misener S; 2000. pp. 365–386. [Google Scholar]
- 37.Southern EM. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}