

Abstract
The objectives of the current studies were to determine the roles of key enzymes in central carbon metabolism in the context of increased production of antibiotics in Streptomyces coelicolor. Genes for glucose-6-phosphate dehydrogenase and phosphoglucomutase (Pgm) were deleted and those for the acetyl coenzyme A carboxylase (ACCase) were overexpressed. Under the conditions tested, glucose-6-phosphate dehydrogenase encoded by zwf2 plays a more important role than that encoded by zwf1 in determining the carbon flux to actinorhodin (Act), while the function of Pgm encoded by SCO7443 is not clearly understood. The pgm-deleted mutant unexpectedly produced abundant glycogen but was impaired in Act production, the exact reverse of what had been anticipated. Overexpression of the ACCase resulted in more rapid utilization of glucose and sharply increased the efficiency of its conversion to Act. From the current experiments, it is concluded that carbon storage metabolism plays a significant role in precursor supply for Act production and that manipulation of central carbohydrate metabolism can lead to an increased production of Act in S. coelicolor.
The wide occurrence of multiply antibiotic-resistant bacterial pathogens of humans has made it urgent to develop new antibiotics. Although over 6,000 different antibiotics have been identified from actinomycetes, these microorganisms are still considered likely to be an important source of further new antibiotics (7). To develop a new antibiotic for clinical application, the compound must be synthesized in sufficient quantities. Random mutation and selection is the tried and tested way to obtain strains producing increased amounts of the targeted compound (11). However, with the advances in genetics, rational or target-directed gene manipulation methods have been developed, and those provide more targeted ways of increasing productivity and discovering novel compounds (12). Such predictive manipulation of central metabolism is likely to be particularly useful at early stages in the testing of new compounds made by genetic manipulation of biosynthetic gene sets in well-characterized surrogate hosts, such as Streptomyces coelicolor or Streptomyces lividans, but the preparation of sufficient quantities of the new compound may be a significant obstacle to further progress.
Antibiotics identified from metabolites of microorganisms are classified into several families, such as polyketides, polyethers, macrolides, and β-lactams, based on chemical structure similarity and common biosynthetic pathways. The provision of intermediates or precursors from primary/intermediary metabolism is a prerequisite for the biosynthesis of secondary metabolites, and the availability of those molecules is a key factor determining the productivity of antibiotics. These precursors are generally formed through the catabolism of various carbon substrates. S. coelicolor produces blue (actinorhodin [Act])- and red (undecylprodiginines [Red])-pigmented antibiotics, which are synthesized at least in part from the same precursors. In Act biosynthesis, on which this paper focuses, seven molecules of acetyl coenzyme A (acetyl-CoA) and one molecule of malonyl-CoA are used to make one molecule of Act by the products of the 23-gene act cluster.
In substrate catabolism, for instance, that of glucose, the Embden-Meyerhof pathway, Entner-Doudoroff pathway, and pentose phosphate pathway (PPP) are interlinked to form a metabolic network. The carbon flux among the pathways is regulated by key enzymes in the individual pathways. Culture conditions, for instance, dissolved oxygen concentration and medium formulation, are very critical determinants of the substrate metabolic rate and flux among the pathways. Glucose-6-phosphate (G6P), the first intermediate in glucose catabolism, is used as a common substrate for phosphoglucose isomerase, glucose-6-phosphate dehydrogenase (Zwf), and phosphoglucomutase (Pgm) (Fig. 1).
FIG. 1.

Central carbon metabolism and intermediates from primary metabolism for Act production in S. coelicolor. G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; F1,6DP, fructose-1,6-diphosphate; GAP, glyceraldehyde-3-phosphate; 1,3 BPG, 1,3-diphosphoglycerate; 3PG, 3-phosphoglycerate; 2PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; 6PGL, 6-phosphoglucolactone; 6PG, 6-phosphogluconate; Ru5P, ribulose-5-phosphate; Ri5P, ribose-5-phosphate; Xu5P, xylulose-5-phosphate; SHu7P, sedoheptulose-7-phosphate; E4P, erythrose-4-phosphate; HK, hexokinase; Pfk, phosphofructokinase; Adl, aldolase; Tpi, triose phosphate isomerase; Gpdh, glyceraldehyde-3-phosphate dehydrogenase; Pgk, phosphoglycerate kinase; Pgm*, phosphoglycerate mutase; Eno, enolase; Pyk, Pyruvate kinase; Pdh, pyruvate dehydrogenase; ACCase, acetyl-CoA carboxylase; Zwf, glucose-6-phosphate dehydrogenase; Pgm, phosphoglucomutase; Pgl, phosphoglucolactonase; Pgdh, phosphogluconate dehydrogenase; Ppi, phosphopentose isomerase; Tkl, transketolase; Tal, transaldolase.
Phosphoglucose isomerase leads carbohydrate catabolism through the Embden-Meyerhof pathway and subsequently to the trichloroacetic acid cycle, where precursors and cofactors for cell growth and also for secondary metabolite formation are provided. The rate of glycolysis and carbon flux into it are determined by phosphofructokinase (Pfk). The PPP is an important pathway providing essential cofactors, such as NADPH, and intermediates for cell growth. Carbon flux into the PPP is determined by Zwf, which has activity during rapid growth (25). Two different genes, zwf1 and zwf2, were identified that encode Zwf in S. lividans, and mutants of S. lividans with deletions of zwf1 or zwf2 showed about four-times-increased production of Act and Red compared to the parent strain, although cell growth of the mutants was not greatly altered (5). On the other hand, a positive correlation between methylenomycin production and carbon flux into the PPP was observed in S. coelicolor (19). Pgm catalyzes the readily reversible interconversion of G1P and G6P, potentially leading to the diversion of carbon to the biosynthesis of glucose-based polymers such as glycogen.
In the current study, we have aimed to reduce the carbon flux into the PPP or the glycogen synthetic pathway by deleting genes for Zwf isozymes and Pgm, respectively. Since acetyl-CoA and/or malonyl-CoA is a precursor for the biosynthesis of Act, a gene complex for acetyl-CoA carboxylase (ACCase), which is the essential enzyme converting acetyl-CoA to malonyl-CoA, was cloned into an expression vector and introduced into the “wild-type” M600 strain of S. coelicolor. Each manipulation had marked effects on Act production, and these could be related to measured changes in the carbon flux from substrate to antibiotic production.
MATERIALS AND METHODS
Bacterial strains, plasmids, and maintenance.
All Streptomyces strains used in this work are listed in Table 1. S. coelicolor M600 and S. coelicolor M510 (ΔredD) derived from S. coelicolor M145 were the source of genomic DNA (16). For maintenance of Streptomyces strains, spores formed on mineral salts agar medium were harvested and suspended in 20% (vol/vol) glycerol, and the spore suspension was stored at −70°C (10). Escherichia coli DH5α was used as a host for the propagation of cosmids and plasmids. E. coli BW25113/pIJ790 was used as a host for PCR-targeted disruption of target genes within cosmid clones (9). E. coli ET12567 (18) was used to obtain methylation-negative DNA. Strains of E. coli were grown in Luria-Bertani medium containing appropriate antibiotics for maintaining plasmids or cosmids. pIJ773 (9) and pIJ774, which is basically the same as pIJ773 but contains a hygromycin resistance gene instead of the apramycin resistance gene, were used for amplification of the apramycin and hygromycin resistance cassettes for the disruption of target genes. Helper plasmid pUZ8002 was used for conjugation from E. coli to Streptomyces.
TABLE 1.
Strain list
Gene manipulation.
DNA manipulation in E. coli and Streptomyces spp. was carried out according to the methods reported elsewhere (26). Enzymes for DNA manipulation were used according to the manufacturer's recommendations (Pos-chem Co. and Boehringer-Manheim). DNA fragments were purified from agarose gel using a gel extraction kit (QIAGEN Co.). Genomic DNA was purified from S. coelicolor M600 using the salting out procedure (22). Transformation with plasmid or cosmid DNA was carried out as described by Kieser et al. (16).
PCR.
Ex-Tag PCR Premix (Takara Inc.) was used in PCR amplification under the following conditions: an initial denaturation step of 2 min at 96°C, 30 cycles of amplification (45 s at 94°C, 45 s at 65°C, 2 min at 72°C), and a final extension period of 15 min at 72°C.
Targeted in-frame deletion of genes.
The genes zwf1 (SCO6661) and zwf2 (SCO1937) were disrupted by PCR targeting using the hygromycin resistance cassette in pIJ774, and pgm (SCO7443) was disrupted using the apramycin resistance cassette in pIJ773 (9). Resistance cassettes were amplified using the High-Fidelity Expand PCR system (Roche Co.) with the primers listed in Table 2. The resistance cassettes were introduced into E. coli BW25113/pIJ790 containing the appropriate cosmid (St5A7, StC54, and St5C11), preinduced for λ RED functions by the addition of arabinose, to obtain a target gene-disrupted version of the mutant cosmid. The disrupted cosmid was isolated and transferred via E. coli ET12567/pUZ8002 to S. coelicolor M510 (ΔredD) by conjugation. Exconjugants were selected on mineral salts agar containing hygromycin (50 μg ml−1) or apramycin (50 μg ml−1), and the products of double crossovers were identified by screening for sensitivity to kanamycin (50 μg ml−1). The disruptions were confirmed by Southern hybridization. [32P]dCTP (Amersham Co.) and the DIG DNA labeling and detection kit (Roche Co.) were used for probe preparation.
TABLE 2.
Primers used for PCR amplification of DNA segments
| Designation | Nucleotide sequence (5′-3′) |
|---|---|
| zwf1-F | ACG AAG TCC CTC GAC AGC AAG GGA GTT GAC GGG GAA TGA TCC CGG GGA TCC GTC GAC C |
| zwf1-R | TGA TCT TGC TTG CCG TGG TGT CGG TCA GGA CGA ACC TCA TGT AGG CTG GAG CTG CTT C |
| zwf1-F | AGC TCA AGC GCC TCG CTC CCT CGA AGG GCT GAG AAT TGA TTC CGG GGA TCC GTC GAC C |
| zwf1-R | GAT CTT GCT GGC CGT GGT GTC CGT GAG GTC GGT CTT CAT TGT AGG CTG GAG CTG CTT C |
| pgm3 | CGA AGT CCC GAC CTA TGC GGA GGA GTG GGC GGC GCA TGA TTC CGG GGA TCC GTC GAC C |
| pgm4 | GCC CGG GGA CCG ACG TAC GGC GTC GGC GCC GCG GTG TCA AGG CTG GAG CTG CTT C |
| ermE1KpnI-F | GAA GGG GGT ACC CAT GCG AGT GTC |
| ermE1-R | GTA ATC GCC GGT CCG CTT CCG GGC |
| accA2KpnI-F | CAC CTT GCG CAC GGT ACC GGC TCC |
| accA2SacI-R | TCA AGG ACT GAT GAG CTC GTC CCG |
| accBEPstI-F | GTA CCT GCA GGA CGG CTC GCA ATC |
| accBEEcoRV-R | CCG AGA TAT CGG GCC TCT CTT GTC |
| accF | TTC GTC AAC GAG ATC AAG CC |
| accR | CAG TCC TTG ATC TCG CAG AT |
Complementation of pgm mutants.
To complement Δpgm mutations, a 4.2-kb EcoRV/XbaI fragment containing pgm and two other genes (SCO7441 and SCO7442) was subcloned from cosmid St5C7 into pUC18 and then excised using EcoRV and XbaI before insertion into similarly cut pSET152s to give pIJ8736 (pSET152s was derived from pSET152, which can integrate efficiently into the chromosomal att site for bacteriophage ΦC31 [3], by replacing the tsr-selective marker with aadA2, encoding spectinomycin resistance). Plasmid pIJ8736 was used to transform Streptomyces strains to spectinomycin resistance.
Cloning and overexpression of ACCase.
To express ACCase strongly and constitutively in S. coelicolor M600, the ermEp* strong promoter (791 bp) was amplified using primers ermE1KpnI-F and ermE1-R. pHCG2 harboring ermEp* as part of a 1.7-kb erythromycin resistance insert was used as template DNA. accA2 was amplified by PCR using primers accA2KpnI-F and accA2ASacI-R. accB and accE were amplified by PCR using accBEPstI-F and accBEEcoRV-R. ermE (791 bp) digested with HindIII/KpnI, accA2 (1,798 bp) digested with KpnI/SacI, and accBE (1,880 bp) digested with PstI/EcoRV were then cloned into pZero-2 (Invitrogen Co.) to give pSMF4701 (3,789 bp), pSMF4702 (5,071 bp), and pSMF4703 (5,154 bp), respectively. A 1,784-bp KpnI/SacI fragment from pSMF4702 was ligated with pSMF4701 digested with the same restriction enzymes to give pSMF4704 (5,563 bp). A 1,868-bp PstI/EcoRV fragment from pSMF4703 was ligated with pSMF4704 digested with the same restriction enzymes to give pSMF4705 (7,420 bp). A 4,225-bp HindIII/XbaI fragment was excised from pSMF4705 and then ligated with pWHM3 (7,200 bp) digested with the same restriction enzymes to give pSMF4706 (11,425 bp) (see Fig. 5). This plasmid has two antibiotic resistance genes (ampicillin/carbenicillin and thiostrepton) and can replicate in either E. coli or Streptomyces because it contains two replication origins (from pUC18 and pIJ101).
FIG. 5.

Construction of pSMF4706 containing genes for the acetyl-CoA carboxylase complex for overexpression in S. coelicolor M600. accA2 (SCO4921) encodes acetyl-CoA carboxylase, and accB (SCO5535) encodes the β-subunit. accE (SCO5536) is thought to help the activity of the acetyl-CoA carboxylase complex.
pSMF4706 was transferred via E. coli ET12567/pUZ8002 to S. coelicolor M600 by protoplast transformation (2), and transformants were selected by overlaying with thiostrepton (50 μg ml−1).
Comparative analysis of acc transcripts from Streptomyces coelicolor.
Spores (108) were germinated in 20 ml of 2× YT (16 g of Bacto tryptone [Difco], 10 g of yeast extract [Difco], and 5 g of NaCl per 1 liter), then harvested by centrifugation, and resuspended in sterile water (3 ml) before 0.1-ml amounts were spread on 8.5-cm cellophane disks on SMMS (28). Mycelium was harvested from several plates every 24 h, then resuspended in 10 ml of RNA protect bacterial reagent (QIAGEN Co.), and incubated for 1 h at 30°C. RNA was purified from the suspension as described in the RNeasy mid kit manual (QIAGEN Co.). First-strand cDNA and double-stranded DNA were synthesized by Superscript II RT (Invitrogen Co.) and Ex-Taq premix (Takara Inc.), respectively. Two primers (accF and accR) were used in PCR (Table 1).
Antibiotic production.
The strains of S. coelicolor used in the fermentor experiments harbored multicopy plasmid pIJ68 carrying the pathway-specific activator gene for Act biosynthesis (actII-ORF4) and the thiostrepton resistance gene (tsr). This plasmid maximized act gene expression so that Act biosynthetic enzymes should not be limiting for Act production. A spore suspension (about 108 spores) stored at −80°C was inoculated into 50-ml of GG1 medium (15 g of glucose, 15 g of glycerol, 15 g of soy peptone, 3 g of NaCl, l g of CaCO3, and 0.05 g of thiostrepton per liter) in 500-ml baffled Erlenmeyer flasks. The first seed cultures were grown for 48 h at 30°C on a rotary shaker at 250 rpm. Then 4 ml of the first seed culture was inoculated into 50 ml of GYB medium (37.5 g of glucose, 15 g of yeast extract, and 0.05 g of thiostrepton per liter) in a 500-ml baffled flask. The second cultures were grown for 24 h at 30°C on a rotary shaker at 250 rpm, and 200 ml of the second seed culture was inoculated into a fermentor containing 2 liters of Evans medium {37.5 g of glucose, 6.6 g of (NH4)2SO4, 0.564 g of NaH2PO4, 0.745 g of KCl, 0.119 g of MgCl2 · 6H2O, 0.284 g of Na2SO4, 0.384 g of citric acid, 0.028 of CaCl2 · 2H2O, 0.0203 g of ZnO, 0.0162 g of FeCl3, 0.00625 g of MnCl2, 0.0067 g of CuCl2, 0.0129 g of CoCl2, 0.00309 g of H3BO3, 0.000021 g of NaMoO4 · 2H2O, and 5.23 g of TES [N-tris (hydroxymethyl)methyl-2-aminoethanesul-phonic acid] per liter of distilled water}. All media were adjusted to pH 7.2 before sterilization at 121°C for 15 min. Glucose, (NH4)2SO4, KH2PO4, NaH2PO4, and other trace elements were sterilized separately and added aseptically. The culture temperature was maintained at 30°C, and pH was adjusted to 7.2 by automatic addition of 1 N HCl or 1 N NaOH. Agitation was fixed at 300 rpm, and aeration was at 1 volume of air per volume of medium per min (vvm).
All fermentation experiments were carried out with duplicate runs. The data shown were averaged from the duplicate fermentations. All fermentation kinetic parameters were calculated with the mean values. The maximum variation was within 5%.
Preparation of samples.
Ten-milliliter samples of cultures were collected aseptically and centrifuged (3,000 × g) at 4°C for 10 min (A-4-81; Eppendorf Co.). Mycelium-free supernatant was used for the determination of glucose and antibiotic concentrations. For the analysis of enzymes, mycelium pellets were resuspended in 3 ml of Tris-HCl (100 mM, pH 7.2) and disrupted by sonication for 3 min (pulse intensity, 25%; pulse on for 5 s and off for 10 s; Uribra cell). Cleared cell extract after centrifugation (15,000 × g, 4°C, 15 min) (5415D; Eppendorf Co.) was used for the determination of enzyme activity.
Analytical methods.
After collection, mycelium was washed twice with physiological saline solution and once with distilled water and dried at 80°C for 24 h for dry weight determination. The glucose concentration was determined using the glucose oxidation method (15). The concentrations of ammonia and phosphate were determined as described elsewhere (8, 21).
The Act concentration was measured as follows (6). First, 0.5 ml of 3 N NaOH was mixed thoroughly with 1 ml of culture sample. After centrifugation (15,000 × g, 2 min), the Act concentration was determined by measuring the absorbance of the supernatant at 640 nm in a UV-160 spectrophotometer (Shimazu Co.), using the molar absorptivity ɛ640 = 25,350 M−1 cm−1. Undecylprodigiosin (Red) was extracted from the cell pellet harvested by centrifugation from 1-ml culture aliquots. An equal volume of methanol (pH 1.0) to culture aliquot was added to the cell pellet and vortexed thoroughly for 2 min. After removing cell debris by centrifugation (maximum speed, 2 min) (5415D; Eppendorf Co.), absorbance measurements were made at 530 nm, and Red concentration was calculated using the molar absorptivity ɛ530 = 100,150 M−1 cm−1 (29).
Glycogen concentration was determined as described in reference 1. Mycelium grown on SMMS (27) was harvested and resuspended in 1 ml of distilled water. Cell disruption was carried out by sonication (6 min, pulse intensity of 25%, pulses were on for 15 s and off for 15 s). Cell-free supernatant was obtained after centrifugation (15,000 × g, 4°C, 15 min). A sample of the supernatant (50 μl) was mixed with 25 μl of sodium acetate buffer (25 mM, pH 4.5) and amyloglucosidase (25 μl) (0.6 units; Sigma Co.) and incubated for 2 h at 37°C. The reaction was stopped by heating at 90°C for 10 min and centrifuged (15,000 × g) for 20 min. The supernatant (10 μl) was mixed with 100 μl of glucose assay reagent (catalog no. G2020; Sigma Co.). After incubation at 25°C for 15 min, the absorbance at 340 nm was measured.
Determination of enzyme activity.
Zwf activity was carried out as reported previously (17). The cell extract (10 μl), 840 μl of Tris-HCl (0.1 M, pH 7.5), and 10 μl of MgCl2 (1 M) were mixed well. After incubation at 30°C for 1 min, 50 μl of NADP+ (0.2 mM) was added and the mixture was incubated at 30°C for 1 min. Ten microliters of glucose-6-phosphate (1 mM) was added, and the rate of change in absorbance was measured at 340 nm.
Pgm activity was determined with a modified method as follows: 50 μl of clear cell extract was mixed with 800 μl of Tris-HCl (0.1 M, pH 7.5), 5 μl of MgCl2 (1 M), 5 μl of glucose-6-phosphate dehydrogenase (0.4 unit), 50 μl of NADP+ (0.2 mM), and 50 μl of glucose-1,6-bisphosphate (1 mM) and incubated at 30°C for 2 min, after which 50 μl of glucose-1-phosphate (1 mM) was added, followed by incubation at 30°C for 1 min. The rate of change in absorbance was measured at 340 nm (17). The protein concentration was measured with a Bradford assay kit (Bio-Rad Co.).
Fermentation kinetic parameters.
Data from duplicate batch cultures were analyzed for specific growth rate (μ), specific rate of glucose uptake (qglu), and specific rates of Act and Red production (qAct and qRed). The fermentation kinetics parameters were analyzed from basic equations (20).
RESULTS
Effect of zwf1 and zwf2 gene deletions on growth and Act production.
This study aimed at evaluating the metabolic effects of zwf deletions in relation to Act production. Two genes, SCO6661 (zwf1) and SCO1937 (zwf2), that encode Zwf were separately deleted by PCR targeting from S. coelicolor M510 (ΔredD) to give mutants M717 (Δred Δzwf1) and M718 (Δred Δzwf2). pIJ68 was introduced into M510, M717, and M718 to maximize expression of the act genes. First, the Zwf activity of the parent strain (M510/pIJ68) and mutants (M717/pIJ68 and M718/pIJ68) was determined with mycelium harvested at multiple time points during batch cultures. Zwf activity in the parent strain was highest during the main growth phase (peaking at 120 h) and decreased greatly in the stationary phase. A closely similar profile was obtained with the Δzwf1 mutant, indicating that zwf1 contributes little of the Zwf activity under these conditions, but the activity was greatly reduced in the Δzwf2 mutant (Fig. 2). In batch culture, glucose consumption, mycelium formation, and Act production in batch cultures were not greatly affected by the deletion of zwf1. However, overall mycelial growth and Act production were enhanced by the deletion of zwf2, while glucose consumption was slightly retarded (Fig. 2). Kinetic parameters calculated from the batch cultures showed that specific glucose uptake rate (qglu) and the specific Act production rate (qAct) were decreased by the deletion of zwf2, while the Act yield based on glucose consumption (YAct/glu) and mycelium formation yield based on glucose consumption (Yx/glu) were apparently increased (Table 3).
FIG. 2.

Changes in Act, dry cell weight (DCW), specific activity of Zwf, and glucose in batch cultures of S. coelicolor M510/pIJ68 (parent type, closed circles), M717/pIJ68 (Δzwf1, open circles), and M718/pIJ68 (Δzwf2, open squares).
TABLE 3.
Comparison of kinetic parameters in derivatives of S. coelicolor
| Strain | Description | Yx/glu (g g−1) | YAct/glu (g g−1) | YAct/x (g g−1) | μ (h−1) | qglu (g g−1 h−1) | qAct (g g−1 h−1) |
|---|---|---|---|---|---|---|---|
| M510/pIJ68 | SCP1−, SCP2−, ΔredD/ermEp*::actII-ORF4 | 0.235 | 0.115 | 0.487 | 0.090 | 0.110 | 9.1 × 10−3 |
| M717/pIJ68 | SCP1−, SCP2−, ΔredDΔzwf1/ermEp*::actII-ORF4 | 0.269 | 0.118 | 0.440 | 0.110 | 0.130 | 6.6 × 10−3 |
| M718/pIJ68 | SCP1−, SCP2−, ΔredD Δzwf2/ermEp*::actII-ORF4 | 0.389 | 0.186 | 0.478 | 0.150 | 0.040 | 6.8 × 10−3 |
| SMF510/pIJ68 | SCP1−, SCP2−, ΔredD Δpgm/ermEp*::actII-ORF4 | 0.332 | 0.067 | 0.203 | 0.137 | 0.083 | 3.0 × 10−3 |
Effects of pgm gene deletion on growth and Act production.
A gene (SCO7443, pgm) predicted to encode Pgm was deleted from M510 by PCR targeting to give mutant SMF510 (Δred Δpgm). The activity of Pgm in these strains was measured in mycelium harvested from liquid culture in YEME (250 rpm, 30°C, 72 h). The Δpgm mutant had only 5.5% of the Pgm activity of the parent strain (M510), and the activity was restored by complementation with a 4.2-kb DNA fragment containing pgm and two adjacent genes from the chromosome, using the integrating plasmid pIJ8736 (data not shown).
On visual inspection, the growth characteristics on SMMS agar were not significantly altered by the deletion of pgm, but Act production was greatly reduced in all Δpgm mutants. (However, the deletion of pgm from the Red+ strain M145 did not eliminate pigmentation, presumably indicating that the deletion had little effect on the production of Red [data not shown].)
To maximize act gene expression, pIJ68 was introduced into SMF510. Batch culture data for glucose consumption, mycelium formation, and Act production in the parent (M510/pIJ68) and the pgm mutant (SMF510/pIJ68) are compared in Fig. 3. Act production was reduced by the deletion of pgm, although mycelium growth was enhanced and the consumption of glucose was almost same. The kinetic values were calculated and showed that Act production rate (qAct) and Act production yield based on glucose consumption (YAct/glu) or on mycelial mass (YAct/x) were reduced (Table 4).
FIG. 3.

Comparison of glucose concentration, dry cell weight (DCW), and Act production in batch cultures of S. coelicolor M510/pIJ68 (parent type, closed circles) and SMF510/pIJ68 (Δpgm, open circles).
TABLE 4.
Effect of overexpressing acetyl-CoA carboxylase on kinetic parameters in S. coelicolor
| Strain | Description | Yx/glu (g g−1) | YAct/glu (g g−1) | YAct/x (g g−1) | μ (h−1) | qglu (g g−1 h−1) | qAct (g g−1 h−1) |
|---|---|---|---|---|---|---|---|
| M600 | SCP1−, SCP2− | 0.483 | 0.012 | 0.028 | 0.077 | 0.030 | 0.6 × 10−3 |
| SMF4703 | SCP1−, SCP2−/ermEp*::accA2::accBE | 0.484 | 0.069 | 0.143 | 0.118 | 0.096 | 5.5 × 10−3 |
In view of the expectation that G1P emanating from the action of Pgm was an essential precursor of glycogen (Fig. 1), the intracellular concentration of glycogen in the parent strain and the SMF510 (Δpgm) mutant was compared (Fig. 4). Remarkably, the mutant made at least as much glycogen as its parent, though the strains differed somewhat in the temporal pattern of accumulation: a possibly growth-phase-dependent periodic fluctuation in glycogen content in the parent strain was absent from the mutant, in which glycogen increased steadily throughout the period of culture.
FIG. 4.
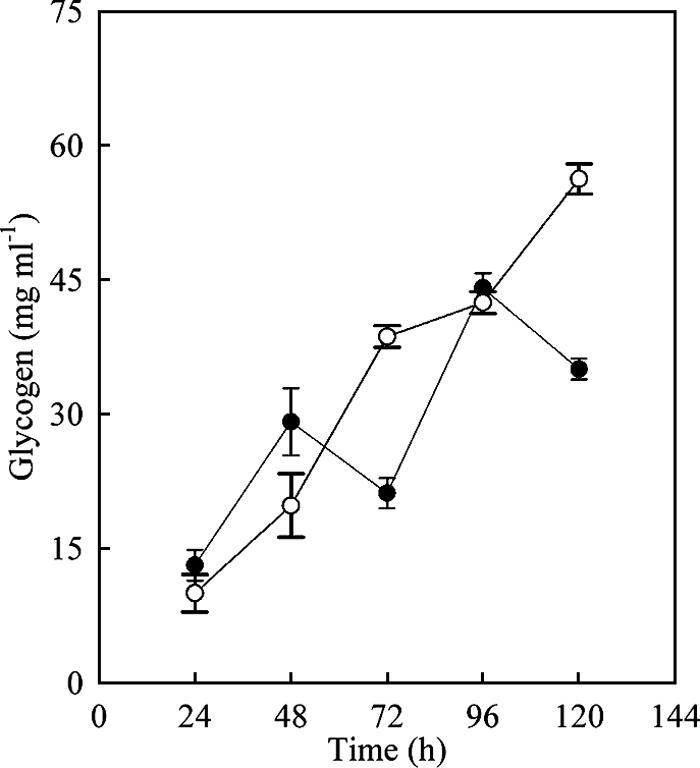
Changes in intracellular glycogen in batch culture of S. coelicolor M510 (parent type, closed circles) and SMF510 (Δpgm) (open circles). Mycelium cultured on the surface of SMMS at 30°C was harvested every 24 h, and cell extracts were used in glycogen assays.
Overexpression of ACCase and antibiotic production.
Malonyl-CoA and methylmalonyl-CoA are the most common chain extender units for the biosynthesis of polyketide antibiotics (13). Therefore, diverting carbon flux from acetyl-CoA away from citrate and directing it toward malonyl-CoA (Fig. 1) should lead to higher antibiotic production. To enhance the carbon flux through acetyl-CoA to malonyl-CoA, the genes encoding the ACCase subunits, accA2, accB, and accE, located in two regions of the genome, were cloned together in an expression vector under the control of a constitutive strong promoter, ermEp* (Fig. 5).
An M600 derivative, SMF4703, harboring the plasmid that contained the acc gene complex was obtained, and it was confirmed that the cloned genes were overexpressed in SMF4703 (Fig. 6). Remarkably, Act production and the glucose uptake in SMF4703 were increased (Fig. 7). Despite this, no specific change in growth was observed. Kinetic parameters showed more clearly that qglu, qAct, YAct/glu, and YAct/x were significantly increased in SMF4703 compared to M600 (Table 4).
FIG. 6.

Reverse transcription-PCR analysis of mRNA transcripts of the ACCase complex. The levels of mRNA transcripts from M600 (parent type) and SMF4703 harboring the ACCase-overexpressing plasmid (pSMF4706) were compared. hrdB was used as a positive control. The samples for purification of mRNA were harvested at mid-exponential phase (M), transition phase (T), and stationary phase (S).
FIG. 7.

Change in concentration of glucose, dry cell weight (DCW), Act, and Red in batch cultures of S. coelicolor M600 (closed symbols) and SMF4703 (with the acc-overexpressing plasmid) (open symbols).
DISCUSSION
We have tried to construct strains producing Act with higher rate and yield by manipulating central carbohydrate metabolism in ways predicted to increase carbon flux from central metabolism to the pathway leading to Act biosynthesis. Although Pfk and pyruvate kinase are the key enzymes regulating central carbohydrate metabolism in many different microorganisms (4, 14), the molecular mechanisms regulating their level and activity in Streptomyces are not known. Instead, we tried to reduce the amount of carbon diverted into either PPP or glycogen biosynthesis by deletion of the zwf or pgm genes.
In studies of S. lividans, it was reported that production of Act and Red was enhanced by the deletion of either zwf gene (zwf1 or zwf2), although mycelium growth was not altered (5). This result indicated that zwf1 and zwf2 contribute equally to carbon flux through PPP in S. lividans. In E. coli, the values for qglu, qCO2, and qace were increased by the deletion of zwf, and the activities of isocitrate dehydrogenase and phosphoglucose isomerase were higher in the Δzwf mutant, indicating that carbon flux though glycolysis and the activities of enzymes involved in the carbon metabolic pathway were enhanced by the deletion of zwf (30).
In the current data on S. coelicolor, it was found that zwf2, rather than zwf1, played the major role in determining Zwf activity. The glucose uptake rate (qglu), Act production rate (qAct), and Act yield based on mycelium formation (YAct/x) were reduced in the Δzwf2 mutant. This means that the overall actinorhodin yield increase could be attributed to the increased growth of the Δzwf2 mutant.
In the second approach, we deleted SCO7443 (pgm), encoding Pgm, which provides the substrate for ADP-glucose pyrophosphorylase, the first enzyme dedicated to glycogen synthesis. Surprisingly, the intracellular concentration of glycogen was, if anything, increased in the mutant (SMF510) and did not show the periodic fluctuation (possibly growth phase associated) of the parent strain (M510) (Fig. 4). These results imply that there is a Pgm-independent route for glycogen synthesis in S. coelicolor. Certainly, there is unusual complexity in the metabolism surrounding the interplay of glycogen and trehalose metabolism in this organism (27). The data in Fig. 4 seem to imply that pgm plays a greater part in glycogen turnover than in its synthesis. Thus, in one explanation for the reduced Act production rate (qAct) and production yield (YAct/glu and YAct/x) of the pgm mutant, carbon stored as glycogen may be a significant source for Act production via Pgm.
Once carbon has been preferentially directed along the glycolytic pathway, some of it is still diverted away from Act when acetyl-CoA is used as the entry molecule to form citrate in trichloroacetic acid, rather than being converted to the Act precursor malonyl-CoA by the ACCase (EC 6.4.1.2). The ACCase complex in S. coelicolor is encoded by three genes, accA1 or accA2, accB, and accE. Among these, accA1 and accA2 have very high nucleotide sequence identity to each other (99%) and accE (196 bp) does not show any identity with other components of ACCase from bacteria but is needed for maximal ACCase activity. In vitro reconstitution of an ACCase revealed that the highest activity of ACCase in heterologous expression was observed from the combination of accA2, accB, and accE (23, 24). To enhance the carbon flux through acetyl-CoA to malonyl-CoA, we incorporated an accA2-, accB-, and accE-overexpressing plasmid into S. coelicolor M600. This led to about a sixfold increase in Act production compared to M600, with qglu and qAct being increased about threefold and eightfold, respectively. From these data, it is concluded that increased production of Act in S. coelicolor is possible by the manipulation of central carbohydrate metabolism. Zwf encoded by zwf2 plays an important role in regulating the carbon flux to Act. It would be interesting to combine the acc and actII-ORF4 expression constructs with deletion of zwf2, but this was not possible with the constructs described here, which were both based on the pIJ101 replicon. The influence of Pgm encoded by SCO7443 is not clearly understood, but the fact that its deletion results in severe reduction in Act production suggests that it would be worth exploring the consequences of pgm overexpression as well as the possibility that glycogen may provide carbon for polyketide biosynthesis.
Acknowledgments
This work was supported by BBSRC grant P14580 and a BK21 Research Fellowship from the Korea Ministry of Education and Human Resource Development.
We are grateful to D. W. Kim for great help on RNA work and to H. J. Hong and A. Hesketh for helpful discussions about the manuscript.
Footnotes
Published ahead of print on 1 September 2006.
REFERENCES
- 1.Bergmeyer, H. U. 1974. Methods of enzymatic analysis, 2nd ed., vol. I., p. 343-435. Academic Press, New York, N.Y. [Google Scholar]
- 2.Bibb, M. J., J. M. Ward, and D. A. Hopwood. 1978. Transformation of plasmid DNA into Streptomyces at high frequency. Nature 274:398-400. [DOI] [PubMed] [Google Scholar]
- 3.Bierman, M., R. Logan, K. O'Brien, E. T. Seno, R. N. Rao, and B. E. Schoner. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43-49. [DOI] [PubMed] [Google Scholar]
- 4.Branny, P., F. De La Torre, and J. R. Garel. 1993. Cloning, sequencing, and expression in Escherichia coli of the gene coding for phosphofructokinase in Lactobacillus bulgaricus. J. Bacteriol. 175:5344-5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler, M. J., P. Bruheim, S. Jovetic, F. Marinelli, P. W. Postma, and M. J. Bibb. 2002. Engineering of primary carbon metabolism for improved antibiotic production in Streptomyces lividans. Appl. Environ. Microbiol. 68:4731-4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bystrykh, L. V., M. A. Fernandez-Moreno, J. K. Herrema, F. Malpartida, D. A. Hopwood, and L. Dijkhuizen. 1996. Production of actinorhodin-related blue pigments by Streptomyces coelicolor A3(2). J. Bacteriol. 178:2238-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chater, K. F. 1990. The improving prospects for yield increase by genetic engineering in antibiotic-producing Streptomycetes. Biotechnology 8:115-121. [DOI] [PubMed] [Google Scholar]
- 8.Fawcett, J. K., and J. E. Scott. 1960. A rapid and precise method for the determination of urea. J. Clin. Pathol. 13:156-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gust, B., G. Chandra, D. Jakimowicz, T. Yuqing, C. J. Bruton, and K. F. Chater. 2004. Lambda red-mediated genetic manipulation of antibiotic producing Streptomyces. Adv. Appl. Microbiol. 54:107-128. [DOI] [PubMed] [Google Scholar]
- 10.Hobbs, G., C. M. Frazer, D. C. J. Gardner, J. A. Cullum, and S. G. Oliver. 1989. Dispersed growth of Streptomyces in liquid culture. Appl. Microbiol. Biotechnol. 31:272-277. [Google Scholar]
- 11.Hopwood, D. A., F. Malpartida, H. M. Kieser, H. Ikeda, J. Duncan, I. Fujii, B. A. Rudd, H. G. Floss, and S. Omura. 1985. Production of ‘hybrid’ antibiotics by genetic engineering. Nature 314:642-644. [DOI] [PubMed] [Google Scholar]
- 12.Hopwood, D. A. 1997. Genetic contributions to understanding polyketide synthases. Chem. Rev. 97:2465-2498. [DOI] [PubMed] [Google Scholar]
- 13.Hopwood, D. A., and D. H. Sherman. 1990. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet. 24:37-66. [DOI] [PubMed] [Google Scholar]
- 14.Kandler, O. 1983. Carbohydrate metabolism in lactic acid bacteria. Antonie Leeuwenhoek 49:209-224. [DOI] [PubMed] [Google Scholar]
- 15.Keston, A. 1956. Specific colorimetric enzymatic reagents for glucose. Abstr. 129th Meet. Am. Chem. Soc., abstr. 31c.
- 16.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom.
- 17.Lessie, T. G., and J. C. Vander Wyk. 1972. Multiple forms of Pseudomonas multivorans glucose-6-phosphate and 6-phosphogluconate dehydrogenases: differences in size, pyridine nucleotide specificity and susceptibility to inhibition by adenosine 5-triphosphate. J. Bacteriol. 110:1107-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacNeil, D. J., K. M. Gewain, C. L. Ruby, G. Dezeny, P. H. Gibbons, and T. MacNeil. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61-68. [DOI] [PubMed] [Google Scholar]
- 19.Obanye, A. I. C., G. Hobbs, D. C. J. Gardner, and S. G. Oliver. 1996. Correlation between carbon flux through the pentose phosphate pathway and production of the antibiotic methylenomycin in Streptomyces coelicolor A3(2). Microbiology 142:133-137. [DOI] [PubMed] [Google Scholar]
- 20.Pirt, S. J. 1975. Principles of microbe and cell cultivation, p. 29-41. Blackwell Scientific Publications, Oxford, United Kingdom.
- 21.Plummer, T. H., Jr., A. W. Phelan, and A. L. Tarentino. 1987. An introduction to practical biochemistry, 3rd ed. McGraw-Hill, London, United Kingdom.
- 22.Pospiech, A., and B. Neumann. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11:213-218. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez, E., and H. Gramajo. 1999. Genetic and biochemical characterization of α- and β-components of a propionyl-CoA carboxylase complex of Streptomyces coelicolor A(3)2. Microbiology 145:3109-3119. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez, E., C. Banchio, L. Diacovich, M. J. Bibb, and H. Gramajo. 2001. Role of an essential acyl coenzyme A carboxylase in the primary and secondary metabolism of Streptomyces coelicolor A3(2). Appl. Environ. Microbiol. 67:4166-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salas, J. S., L. M. Quiros, and C. Hardisson. 1984. Pathways of glucose catabolism during germination of Streptomyces spores. FEMS Microbiol. Lett. 22:229-233. [Google Scholar]
- 26.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 27.Schneider, D., C. J. Bruton, and K. F.Chater. 2000. Duplicated gene clusters suggest an interplay of glycogen and trehalose metabolism during sequential stages of aerial mycelium development in Streptomyces coelicolor A3(2). Mol. Gen. Genet. 263:543-553. [DOI] [PubMed] [Google Scholar]
- 28.Strauch, E., E. Takano, H. A. Baylis, and M. J. Bibb. 1991. The stringent response in Streptomyces coelicolor A3(2). Mol. Microbiol. 5:289-298. [DOI] [PubMed] [Google Scholar]
- 29.Williams, R. P., J. A. Green, and D. A. Rppoport. 1956. Studies on pigmentation of Serratia marcescens. 1. Spectral and paper chromatographic properties of prodigiosin. J. Bacteriol. 71:115-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao, J., T. Baba, H. Mori, and K. Shimizu. 2004. Global metabolic response of Escherichia coli to gnd or zwf gene-knockout, based on 13C-labeling experiments and the measurement of enzyme activities. Appl. Microbiol. Biotechnol. 64:91-98. [DOI] [PubMed] [Google Scholar]