

Abstract
A molecular assembly based on derivatized polystyrene is described, which mimics both the light-harvesting and energy-conversion steps of photosynthesis. The system is unique in that the two key parts of a photosynthetic system are incorporated in a functional assembly constructed from polypyridine complexes of RuII. This system is truly artificial, as none of the components used in construction of the assembly are present in a natural photosynthetic system. Quantitative evaluation of the energy and electron transfer dynamics after transient irradiation by visible light offers important insights into the mechanisms of energy transport and electron transfer that lead to photosynthetic light-to-chemical energy conversion.
In natural photosynthesis, light is converted into chemical energy by a sequence of coupled energy and electron transfer events. Initially, sunlight is absorbed by light-harvesting pigments, chlorophylls and carotenoids, which act as antenna fragments. The energy collected by absorption creates molecular excited states and is subsequently transferred between chromophores through an energy cascade ultimately reaching the reaction center. At the reaction center, the light energy is converted into chemical energy by a series of electron transfer steps (1–5). Understanding of these concepts has motivated chemists to design assemblies that have related properties and may provide the basis for artificial solar energy conversion devices (6–10). The construction of a functional and efficient artificial solar energy conversion device would have a significant impact on our ability to use solar energy.
To date, most research on the artificial solar energy conversion has focused on mimicking various aspects of natural photosynthesis. Through these studies, a reasonable understanding of various components necessary for construction of an artificial photosynthetic system has been developed. The main challenge at the moment is to assemble these components in a spatially organized manner, which will allow their efficient collaboration in harvesting the light energy and converting it efficiently into chemical energy. There are currently two strategies. One is based on constructing complex supramolecular assemblies in which the various components are linked together by chemical bonds (11–22). The second utilizes various supports such as polymers (23–32), zeolites (33–38), sol-gel glasses (39–41), lipid membranes (42, 43), or self-assembled films (44–47) as scaffolds for incorporating and organizing the necessary components.
Derivatized polymers provide an attractive approach to this problem because they offer flexibility and simplicity in the design of multifunctional assemblies. We (23–28) and others (29–32) have used derivatized polymers to gain insight into various aspects of the photosynthetic process. Recently, we showed, for example, that polymeric assemblies derivatized with polypyridyl complexes of RuII and OsII display antenna and energy transport properties similar to natural photosynthetic systems (48, 49). In the present work, we describe an assembly that combines both the light-harvesting and electron-transfer properties of a natural photosynthetic system within a single macromolecule.
The support of choice in these studies was a mixed styrene-based copolymer abbreviated as [co-PS-CH2Cl], which was prepared by free radical random copolymerization of styrene with p-(chloromethyl)styrene by standard methods (50, 51). This copolymer is a versatile precursor for the addition of a variety of functional groups. Nucleophilic displacement of chloride from the pendant chloromethyl groups provides a means for attachment of functional groups by either ether or amide linkages (23–28).
The amide-linked polymers were prepared by displacement of the chloride
group by potassium phthalimide, followed by reduction with hydrazine to
give the amine-functionalized polymer,
[co-PS-CH2NH2].
This derivatization allows transition metal complexes derivatized with
carboxylic acid groups such as
[M(bpy)2(bpyCOOH)]2+
(M = RuII or OsII, bpy
is 2,2′-bipyridine, and bpyCOOH is 4-methyl-4′-carboxy-2,2′-bipyridine)
to be attached by using standard amide-coupling procedures (51, 52).
The structure of a repeat unit of the resulting polymer 1 is
illustrated below.
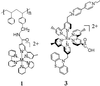
By controlling the amount and order of addition, it is possible to attach more than one complex on single polymer strand, allowing for the construction of complex molecular assemblies with tailored properties (23–25, 28).
In the mixed polymer [co-PS-CH2NHCO-(Os3IIRu13II)](PF6)32 (2), prepared by attachment of [OsII(bpy)2(bpyCOOH)]2+ and [RuII(bpy)2(bpyCOOH)]2+ to the amine parent polymer, RuII→polypyridine excitation at the high energy majority RuII sites creates a metal-to-ligand charge transfer (MLCT) excited state of energy 2.13 eV (in CH3CN solvent). Formation of the excited state is followed by energy transfer to the lower energy OsII trap sites to give OsII* (1.77 eV) (48). Energy transfer is rapid (k ≈1 × 109 s−1) and efficient (η = 0.98).
In the current series of experiments, we replaced the OsII trap with the reaction center model [RuII(bpyCOOH)(bpyCH2PTZ)(bpyCH2MV2+)]4+ (RC, 3) (53). It is both a MLCT chromophore and a chromophore-quencher complex with the electron transfer donor [phenothiazine (PTZ)] and acceptor [methylviologen (MV2+)] appended to metal-bound ligands. After RuII → polypyridine excitation at 460 nm in CH3CN, MLCT, excited-state energy within the complex is converted into transient chemical energy by a series of electron transfer steps. This gives a redox-separated (RS) state with transiently stored oxidative, −PTZ+. and reductive, − MV+. redox equivalents with an efficiency of ηRS = 0.35 ± 0.05. The RS state decays to the ground state by back electron transfer with kb = 6.3 × 106 s−1(t = 160 ns) (53).
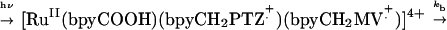 |
1 |
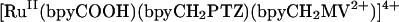 |
The mixed polymer was prepared by addition of three equivalents (out of 20) of 3 followed by an excess of [RuII(bpy)2(bpyCOOH)]2+ (18). This gave the mixed polymer assembly, [co-PS-CH2NHCO-(RC3Ru17II)](PF6)46 (4), a fragment of which is illustrated in Fig. 1.
Figure 1.

(a) A kinetic scheme illustrating the antenna effect in antenna/reaction center polymer, 4. Irradiation of the assembly by visible light leads to the excitation of a bound chromophore unit, [RuII(bpy)2(bpyCONH-)](PF6)2, which acts as an antenna fragment within the polymer. After absorption of a photon and thermal equilibration, a 2.13-eV excited state is formed, which can rapidly transfer its energy to a neighbor. In this way, the energy migrates along the polymer chain until it reaches the reaction center model [RuII(bpyCONH-) (bpyCH2PTZ)(bpyCH2MV2+)](PF6)4. The MLCT state at the reaction center is formed by energy transfer, and it initializes stepwise intramolecular electron transfer to form a RS state at 1.15 eV. The RS state subsequently undergoes back electron transfer with k(CH3CN, 25°C) = 6.3 × 106 s−1. (b) A kinetic scheme explaining the appearance of the long-lived component. In this case the antenna excited state undergoes electron transfer to MV2+ acting as acceptor. The electron hole generated in this way migrates along the polymer chain by RuII→RuIII electron self exchange (migration) leading to spatially isolated −PTZ+. and −MV+. sites. The same intermediate could form after electron donation by PTZ followed by electron migration.
In 4, the MLCT excited-state energy of the chromophore-quencher sites is 2.04 eV (53), so that energy transfer after excitation at the majority antenna sites is favored by −0.09 eV. Energy transfer is manifested by a decrease in emission quantum yield (φem = 0.037) compared with the model homopolymer [co-PS-CH2NHCO-(Ru16II)](PF6)32 (5) (φem = 0.064). Excited state quenching is an intramolecular effect independent of sample concentration. The quenching efficiency is 34% on excitation at 460 nm (in CH3CN at 25°C) after correcting for the light absorbed by the reaction center fragment.
The origin of the quenching was demonstrated by laser flash photolysis with transient absorption monitoring in CH3CN at 25°C. Transient absorption difference spectra are shown in Fig. 2 for polymer assembly 4, homopolymer 5, and unbound chromophore-quencher assembly 3. The characteristic features for 3 are the appearance of absorption bands for reduced MV2+ at 398 and 608 nm (54) and oxidized PTZ at 517 nm (55). The difference spectrum of 4 is more complex because of an overlapping contribution from unquenched antenna excited states, which have a characteristic absorption at 380 nm and a bleach at ≈460 nm.
Figure 2.

Transient absorbance difference spectra for (a) homopolymer [co-PS-CH2NHCO-(RuII16)](PF6)32 (5) and the assembly [co-PS-CH2NHCO-(RuII(RC)3RuII17)](PF6)46 (4) and (b) the unbound chromophore-quencher assembly [RuII(bpyCOOH)(bpyCH2PTZ)(bpyCH2MV2+)](PF6)4 (3) in CH3CN at 25°C. The spectra were obtained 100 ns after 460 nm excitation (≈7 ns FWHM), 1.82 mJ/pulse cm2 from a Nd:YAG/OPO laser system with the excitation beam defocused to 3.85 cm2. The spectra in a are corrected for a difference in ground state absorbance.
The separate decay processes were deconvoluted by kinetic monitoring at 401 nm, which is both an isosbestic point for the antenna excited state and a wavelength of significant absorption for MV+., and at 450 nm, where the absorption change is dominated by the MLCT ground-state bleach. The results are shown in Fig. 3 as absorbance (A) − time (t) traces. The important observations are: (i) The presence of both short- and long-lived components in 4 with monitoring at 401 nm (Fig. 3a); (ii) the differences between ΔAmax (4) and ΔAmax (5), both at 450 nm and 401 nm; and (iii) the difference in decay kinetics of 4 and 5 at 450 nm (Fig. 3b).
Figure 3.

(a) Comparison of absorbance (ΔA) − time (t) changes in the assembly [co-PS-CH2NHCO-(RuII(RC)3RuII17)](PF6)46 (4) and homopolymer [co-PS-CH2NHCO-(RuII16)](PF6)32 (5). At the monitoring wavelength of 401 nm, unquenched antenna excited states do not contribute to the observed signal. The observed absorbance change is associated with the appearance of MV+. in the RS state. The large value of ΔAmax shows that a considerable fraction of the RS state forms during the laser pulse. There is also clear evidence for both rapid and slow decay processes after the laser pulse. Experimental conditions as in Fig. 2. (b) Transient absorbance change at the bleach (450 nm) obtained after 500 nm excitation, 1.82 mJ/pulse⋅cm2 with the excitation beam defocused to 3.85 cm2. The smaller ΔAmax change for 4 compared with 5 is consistent with a rapid quenching process or processes that occur within the laser flash. The enhanced decay kinetics for 4 compared with 5 occur because of energy migration and quenching by sensitization of the RC. Fitting the decay traces to Eq. 3 gives for 4 k1 = 6.7 × 107 s−1 (ΔA1 = 10.10 mOD), k2 = 9.0 × 106 s−1 (ΔA2 = 8.47 mOD), k3 = 1.3 × 106 s−1 (ΔA3 = 6.67 mOD) and for 5 k1 = 3.7 × 107 s−1 (ΔA1 = 14.08 mOD), k2 = 3.8 × 106 s−1 (ΔA2 = 8.42 mOD), k3 = 7.8 × 105 s−1 (ΔA3 = 10.91 mOD). With use of Eq. 4, this gives the average decay rate constants <k (4)> = 4 × 106 s−1 and <k (5)> = 2 × 106 s−1.
From the ΔAmax change at 401 nm, a significant amount of the RS state on the polymer forms during the ≈7-ns pulse. This is consistent with the decrease in ΔAmax for 4 compared with 5 observed at 450 nm. By accounting for the fraction of light absorbed by the reaction center, and from the difference in ΔAmax (4) and ΔAmax (5) at multiple monitoring wavelengths, it can be estimated that the rapid formation of the RS state is partially, ≈50%, because of direct excitation and electron transfer at the RC and partially, ≈50%, because of excitation at antenna sites followed by energy transfer and sensitization. In interpreting these results, it is important to recognize that, as illustrated in Eq. 2, there are different antenna sites, those adjacent to the RC (RuaII) and those at least once removed (RubII). Fast formation of the RS state is associated with both direct excitation of the RC and excitation at RuaII followed by rapid energy transfer (ken in Eq. 2),
 |
2 |
which, as noted above, is favored by ΔG° = −0.09 eV.
Sensitization after excitation at RubII requires an initial energy migration step (kmig in Eq. 2) before energy transfer can occur. Energy migration followed by energy transfer and sensitization accounts for the faster recovery of the MLCT ground state bleach in 4 compared with 5 (Fig. 3b).
For 4 and 5, absorbance changes with time, ΔA(t), at 450 nm are kinetically complex but could be fit to Eq. 3,
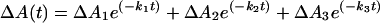 |
3 |
with an average rate constant calculated from Eq. 4.
 |
4 |
Analysis of the data in Fig. 3b gives for 4 <k (4)> = 4.0 (±0.2) × 106 s−1 and for 5 <k (5)> = 2.0(±0.1) × 106 s−1. The average quenching rate constant, <kq> = 2.0(±0.2) × 106 s−1, can be calculated from the relationship <kq> = <k (4)> − <k (5)>. On the basis of this value and <k (5)>, the quenching efficiency after excitation at RubII is
 |
, which shows that, after excitation, approximately 1/2 of the excited RubII sites are effective in sensitizing the formation of the RS state.
Quantitative analysis of the kinetic decay trace in Fig. 3a is complicated by the appearance of a small nonzero ΔA change that persists even after ≈10 μs. In an independent experiment, it was shown that this long-lived component in fact persists for 20.8 ms and decays by second-order equal-concentration kinetics with k ≈ 48 M−1⋅s−1. An explanation for this behavior is suggested in Fig. 1b, which invokes competitive quenching of an antenna site by electron transfer to MV2+, followed by hole migration via RuIII →RuII electron transfer. Initial PTZ quenching followed by electron migration would give the same result with oxidative and reductive equivalents trapped on spatially separated sites. In this case, intrastrand back-electron transfer is slow (k ≲ 15 s−1), and the mechanism is diffusional with back electron transfer occurring between −MV+. and −PTZ+. on separate strands.
The transient absorption results demonstrate that assembly 4 functions as an antenna for the sensitization of electron transfer. Sensitized electron transfer occurs by a combination of direct excitation of the reaction center fragment and energy transfer and energy migration followed by sensitized electron transfer. The efficiency of sensitized electron transfer after excitation of an antenna fragment (ηsen) can be calculated by using Eq. 5 and chromophore-quencher complex 3 as a photon counter.
 |
5 |
In Eq. 5, ΔA(3)∫ is the time-integrated transient absorbance signal at 401 nm for 3, ΔA(4)∫ is the same for 4 (excluding contributions from the long-lived component and direct excitation), η(3)RS is the quantum yield for the formation of the RS state in 3 (η(3)RS = 0.35) (53), and F(4) is the fraction of light absorbed by antenna molecules RuaII and RubII. On the basis of this analysis and the data in Fig. 3a, at an excitation irradiance of 1.8 mJ/pulse cm2, ηsen = 12%. Under the same conditions, the yield for the long-lived transient is ≈0.5%.
The sensitization efficiency is irradiance dependent, with ηsen decreasing from 17.5% at 0.22 mJ/pulse cm2 to 11.5% at 2.39 mJ/pulse cm2 (Fig. 4). The higher value is consistent with the quenching yield determined by steady-state measurements under conditions of low incident irradiance, because ηsen = Φqη(3)RSS = 17.5%.
Figure 4.

Excitation irradiance dependence of the average excited state lifetime <τ> of the homopolymer [co-PS-CH2NHCO-(RuII16)](PF6)32 (5) (in CH3CN at 25°C) overlaid with the excitation irradiance dependence for the formation of the redox separated state ηsen in antenna-reaction center assembly [co-PS-CH2NHCO-(RuII(RC)3RuII17)](PF6)46 (4). Values for ηsen were calculated by using Eq. 5. The decrease in both quantities with increasing irradiance is a consequence of multiphoton excitation followed by self quenching within individual polymeric strands.
The irradiance dependence can be explained by the existence of multiphoton effects previously identified in related homopolymers (26, 27). At high photon fluxes, multiple excited states are produced on a single strand, and they undergo self quenching. This creates an additional intrastrand mechanism for excited-state decay and decreases the efficiency of RS state formation.
On the basis of our results, the 2.13 eV of excited state energy collected in the antenna fragment of 4 after visible light absorption and equilibration is transferred to a reaction center where it is converted into spatially separated redox equivalents as a 1.15-eV RS state. The overall efficiency is 12–18% depending on irradiance. This efficiency is lower than those obtained recently in some more complex systems (12, 47). However, our results are important in showing that a functional model for the light-harvesting sensitized electron-transfer part of the natural photosynthetic apparatus can be constructed by using polypyridine complexes of RuII without the use of any component that is a part of a natural photosynthetic system. The polymer system reported here is a first generation result, far from being optimized. There are two obvious routes for improvement in later generations of assemblies. One is to increase the driving force for energy transfer from the antenna chromophores to the RC to increase the energy transfer efficiency to the >90% efficiency observed for RuII-OsII polymer 2. The other is to identify RC models for which the redox separation efficiency is greater than 35%. Finally, there are well established routes for immobilizing these polystyrene-based polymer assemblies on conducting supports (56, 57) or in sol-gel matrices (58, 59) for possible device applications.
Acknowledgments
This work was supported by funding from the Division of Chemical Sciences Office of Basic Energy Sciences, U. S. Department of Energy (Grant No. DE-FG02–96ER14607). We thank Dr. Laurence M. Dupray for providing a sample of precursor styrene copolymer.
Abbreviations
- MLCT
metal-to-ligand charge transfer
- PTZ
phenothiazine
- MV2+
methylviologen
- RS
redox separated
References
- 1.Govindjee . Photosynthesis. Energy Conversion by Plants and Bacteria. New York: Academic; 1982. [Google Scholar]
- 2.Danks S M, Evans E H, Whittaker P A. Photosynthetic Systems: Structure, Function and Assembly. New York: Wiley; 1983. [Google Scholar]
- 3.Kuhlbrandt W, Neng Wang D, Fujiyoshi Y. Nature (London) 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 4.Kuhlbrandt W. Structure (Cambridge, UK) 1995;3:521–525. doi: 10.1016/s0969-2126(01)00184-8. [DOI] [PubMed] [Google Scholar]
- 5.Norris J R, Schiffer M. Chem. Eng. News. 1990. 22–37. [Google Scholar]
- 6.Freemantle M. Chem Eng News. 1998;76:37–45. [Google Scholar]
- 7.Meyer T J. Acc Chem Res. 1989;22:163–170. [Google Scholar]
- 8.Norris J R, Jr, Meisel D. Photochemical Energy Conversion. New York: Elsevier; 1988. [Google Scholar]
- 9.Conoly J S. Photochemical Conversion and Storage of Solar Energy. New York: Academic; 1981. [Google Scholar]
- 10.Gust D, Moore T A, Moore A L. Pure Appl Chem. 1998;70:2189–2200. [Google Scholar]
- 11.Gust D, Moore T A, Moore A L, MacPherson A N, Lopez A, DeGraziano J M, Gouni I, Bittersman E, Seely G R, Gao F, et al. J Am Chem Soc. 1993;115:11141–11152. [Google Scholar]
- 12.Gust D, Moore T A, Moore A L, Lee S J, Bittersmann E, Luttrull D K, Rehms A A, DeGraziano J M, Ma X C, Gao F, et al. Science. 1990;248:199–201. doi: 10.1126/science.248.4952.199. [DOI] [PubMed] [Google Scholar]
- 13.Gust D, Moore T A, Moore A L. Acc Chem Res. 1993;26:198–205. [Google Scholar]
- 14.Gust D, Moore T A. Science. 1989;244:35–41. doi: 10.1126/science.244.4900.35. [DOI] [PubMed] [Google Scholar]
- 15.Kuciauskas D, Liddell P A, Lin S, Johnson T E, Weghorn S J, Lindsey J S, Moore A L, Moore T A, Gust D. J Am Chem Soc. 1999;121:8604–8614. [Google Scholar]
- 16.Wagner R W, Johnson T E, Lindsey J S. J Am Chem Soc. 1996;118:11166–11180. [Google Scholar]
- 17.Hsiao J-S, Krueger B P, Wagner R W, Johnson T E, Delaney J K, Mauzerall D C, Fleming G R, Lindsey J S, Bocian D F, Donohoe R J. J Am Chem Soc. 1996;120:11181–11193. [Google Scholar]
- 18.Wasielewski M R. Chem Rev. 1992;92:435–461. [Google Scholar]
- 19.Vogtle F, Plevoets M, Nieger M, Azzellini G C, Credi A, De Cola L, De Marchis V, Venturi M, Balzani V. J Am Chem Soc. 1999;121:6290–6298. [Google Scholar]
- 20.Balzani V, Campagna S, Denti G, Juris A, Serroni S, Venturi M. Acc Chem Res. 1998;31:26–34. [Google Scholar]
- 21.Stewart G M, Fox M A. J Am Chem Soc. 1996;118:4354–4360. [Google Scholar]
- 22.Venturi M, Serroni S, Juris A, Campagna S, Balzani V. Dendrimers. Vol. 197. Berlin: Springer; 1998. pp. 193–228. [Google Scholar]
- 23.Baxter S M, Jones W E J, Danielson E, Worl L, Strouse G, Younathan J, Meyer T J. Coord Chem Rev. 1991;111:47–71. [Google Scholar]
- 24.Younathan J N, Jones W E, Meyer T J. J Phys Chem. 1991;95:488–492. [Google Scholar]
- 25.Strouse G F, Worl L A, Younathan J N, Meyer T J. J Am Chem Soc. 1989;111:9101–9102. [Google Scholar]
- 26.Worl L A, Jones W E, Strouse G F, Younathan J N, Danielson E, Maxwell K A, Sykora M, Meyer T J. Inorg Chem. 1999;38:2705–2708. [Google Scholar]
- 27.Worl L A, Strouse G F, Younathan J N, Baxter S M, Meyer T J. J Am Chem Soc. 1990;112:7571–7578. [Google Scholar]
- 28.Olmsted J I, McClanahan S F, Danielson E, Younathan J N, Meyer T J. J Am Chem Soc. 1987;109:3297–3301. [Google Scholar]
- 29.Fossum R D, Fox M A. J Am Chem Soc. 1997;119:1197–1207. [Google Scholar]
- 30.Fox M A. Acc Chem Res. 1992;25:569–574. [Google Scholar]
- 31.Watkins D M, Fox M A. J Am Chem Soc. 1996;118:4344–4353. [Google Scholar]
- 32.Whitesell J K, Chang H K, Fox M A, Galoppini E, Watkins D M, Fox H, Hong B. Pure Appl Chem. 1996;68:1469–1474. [Google Scholar]
- 33.Borja M, Dutta P K. Nature (London) 1993;362:43–45. [Google Scholar]
- 34.Dutta P K, Ledney M. In: Prog. Inorg. Chem. Karlin K D, editor. Vol. 44. NewYork: Wiley; 1997. pp. 209–271. [Google Scholar]
- 35.Sykora M, Kincaid J R. Nature (London) 1997;387:162–164. [Google Scholar]
- 36.Sykora M, Maruszewski K, Treffert-Ziemelis S M, Kincaid J R. J Am Chem Soc. 1998;120:3490–3498. [Google Scholar]
- 37.Krueger J S, Mayer J E, Mallouk T E. J Am Chem Soc. 1988;110:8232–8234. [Google Scholar]
- 38.Ledney M, Dutta P K. J Am Chem Soc. 1995;117:7687–7695. [Google Scholar]
- 39.Slama-Schwok A, Avnir D, Ottolenghi M. J Am Chem Soc. 1991;113:3984–3985. [Google Scholar]
- 40.Slama-Schwok A, Avnir D, Ottolenghi M. Nature (London) 1992;335:240–242. [Google Scholar]
- 41.Castellano F N, Meyer G J. J Phys Chem. 1995;99:14742–14748. [Google Scholar]
- 42.Steinberg-Yfrach G, Liddell P A, Hung S-C, Moore A L, Gust D, Moore T A. Nature (London) 1997;385:239–241. [Google Scholar]
- 43.Steinberg-Yfrach G, Rigaud J L, Durantini E N, Moore A L, Gust D, Moore T A. Nature (London) 1998;392:479–482. doi: 10.1038/33116. [DOI] [PubMed] [Google Scholar]
- 44.Vermeulen L A, Thompson M E. Nature (London) 1992;358:656–658. [Google Scholar]
- 45.Byrd H, Suponeva E P, Bocarsly A B, Thompson M E. Nature (London) 1996;380:610–612. [Google Scholar]
- 46.Kaschak D M, Johnson S A, Waraksa C C, Pogue J, Mallouk T E. Coord Chem Rev. 1999;186:403–416. [Google Scholar]
- 47.Kaschak D M, Lean J T, Waraksa C C, Saupe G B, Usami H, Mallouk T E. J Am Chem Soc. 1999;121:3435–3445. [Google Scholar]
- 48.Dupray L M, Devenney M, Striplin D R, Meyer T J. J Am Chem Soc. 1997;119:10243–10244. [Google Scholar]
- 49.Jones W E J, Baxter S M, Strouse G F, Meyer T J. J Am Chem Soc. 1993;115:7363–7373. [Google Scholar]
- 50.Arshady R, Reddy B S R, George M H. Polymer. 1984;25:716–721. [Google Scholar]
- 51.Dupray L, Meyer T J. Inorg Chem. 1996;35:6299–6307. [Google Scholar]
- 52.Friesen D, Kajita T, Danielson E, Meyer T J. Inorg Chem. 1998;37:2756–2762. doi: 10.1021/ic9710420. [DOI] [PubMed] [Google Scholar]
- 53.Maxwell K A, Sykora M, DeSimone J M, Meyer T J. Inorg Chem. 2000;39:71–75. doi: 10.1021/ic990512i. [DOI] [PubMed] [Google Scholar]
- 54.Watanabe T, Honda K. J Phys Chem. 1982;86:2617–2619. [Google Scholar]
- 55.Hester R E, Williams K P J. J Chem Soc Perkins Trans. 1981;2:852–859. [Google Scholar]
- 56.Kajita T, Leasure R M, Devenney M, Friesen D, Meyer T J. Inorg. Chem. 1998. 4782–4794. [DOI] [PubMed] [Google Scholar]
- 57.Leasure R M, Kajita T, Meyer T J. Inorg Chem. 1996;35:5962–5963. [Google Scholar]
- 58.Sykora M, Meyer T J. Chem Mat. 1999;11:1186–1189. [Google Scholar]
- 59.Sykora M, Maxwell K A, Meyer T J. Inorg Chem. 1999;38:3596–3597. doi: 10.1021/ic990054d. [DOI] [PubMed] [Google Scholar]