

Abstract
Understanding the evolutionary dynamics of influenza A virus is central to its surveillance and control. While immune-driven antigenic drift is a key determinant of viral evolution across epidemic seasons, the evolutionary processes shaping influenza virus diversity within seasons are less clear. Here we show with a phylogenetic analysis of 413 complete genomes of human H3N2 influenza A viruses collected between 1997 and 2005 from New York State, United States, that genetic diversity is both abundant and largely generated through the seasonal importation of multiple divergent clades of the same subtype. These clades cocirculated within New York State, allowing frequent reassortment and generating genome-wide diversity. However, relatively low levels of positive selection and genetic diversity were observed at amino acid sites considered important in antigenic drift. These results indicate that adaptive evolution occurs only sporadically in influenza A virus; rather, the stochastic processes of viral migration and clade reassortment play a vital role in shaping short-term evolutionary dynamics. Thus, predicting future patterns of influenza virus evolution for vaccine strain selection is inherently complex and requires intensive surveillance, whole-genome sequencing, and phenotypic analysis.
Synopsis
A comparative analysis of 413 complete genomes of the H3N2 subtype of human influenza A virus sampled from New York State, United States, the largest and first population-based study of its kind, reveals that viral evolution within epidemic seasons is dominated by the random importation of genetically different viral strains from other geographic areas, rather than by natural selection favoring local strains able to escape the human immune response. Multiple clades of genetically distinct viral strains cocirculate across the entire state within any season and occasionally exchange genes through reassortment. Both genetic diversity and geographic viral “traffic” are extensive within epidemics. Therefore, the evolution of influenza A virus is strongly shaped by random migration and reassortment and is far harder to predict than previously realized. Consequently, intensive sampling and whole-genome sequencing are essential for selecting viral strains for future vaccine production.
Introduction
The antigenic drift of human influenza A virus is thought to reflect the continuous fixation of advantageous mutations in the surface hemagglutinin (HA) glycoprotein that confer escape from residual host antibody responses [1–3]. This process is depicted in phylogenetic trees of the highly antigenic hemagglutinin HA1 domain in A(H3N2) subtype viruses, the dominant subtype circulating in humans since 1968 [4–6]. These phylogenies are characterized by a single “trunk” lineage that represents the temporal sequence of immune escape variants across epidemic seasons. Viral isolates falling on short side branches persist only briefly (average of ∼1.6 y), reflecting their lower fitness due to a lack of antigenic novelty [3,5].
Our current understanding of antigenic drift is based upon a spatially and temporally thin sampling of HA1 sequences; most were collected by World Health Organization reference laboratories for vaccine strain selection and tend to be preselected for particular virulence or unusual characteristics [7,8]. While vaccine strain selection is based on extensive sampling of viral antigenic properties and supplemented by sequence information, significant vaccine-epidemic mismatches occur on occasion: for example, in the 1997–1998 season, when Sydney-type viruses replaced the dominant Wuhan-type strains [9–10], and in 2003–2004, when Fujian-like viruses replaced Sydney strains [11]. These mismatches are evidently a consequence of the evolutionary plasticity of the HA gene: rather than experiencing a constant rate of antigenic change, HA evolution is characterized by sporadic jumps in antigenic space, generating new antigenic clusters that mismatch the vaccine in use [12]. In some cases these antigenic jumps arise from intrahuman reassortment [13], including among isolates of the same subtype [14,15].
It is therefore critical to determine the processes involved in the genesis of genetic and antigenic diversity, which we hoped to observe within a discrete population over the course of seven intensively sampled influenza seasons. To this end, we performed the largest analysis to date of viral evolution in a spatially and temporally restricted setting, comprising 413 influenza A(H3N2) whole-genome sequences from viruses sampled from 52 of 62 counties in New York State from 1997–2005, supplemented by a dataset of global influenza isolates. These data provide a relatively representative picture of influenza activity in both rural and more urban areas, given that relatively few isolates were sampled from New York City.
Results
Although we explored the evolution of each viral segment, we focused our analysis on the HA and neuraminidase (NA) glycoprotein-encoding genes, for which a larger sample of global background isolates is available. Further, a phylogenetic tree of the six concatenated internal genes of the New York State isolates closely resembled the topological structure of the NA tree (Figure S1). Our phylogenetic analysis revealed substantial genetic diversity in the NA and HA genes within each epidemic season, exemplified by multiple cocirculating clades containing viral isolates both from within New York State and other locations worldwide (Figures 1 and 2; phylogenetic trees for the NA, HA, and concatenated internal without colour labeling are shown in Figures S2, S3, and S4, respectively). The most parsimonious explanation for this pattern, particularly given the relatively small sample of global “background” isolates, is that each clade represents a novel introduction event of a genetically distinct influenza isolate into New York State. In addition, major incongruities between trees inferred for the HA, NA, and concatenated internal gene segments indicate that genomic reassortment events occurred between these cocirculating clades.
Figure 1. Phylogenetic Relationships of the NA Gene of Influenza Viruses Sampled from New York State and Globally during 1997–2005, Estimated Using an ML Method.

Rectangles represent clusters of related New York State isolates, with the size of the rectangle reflecting the number of isolates in the clade. Roman numerals indicate separate clades, with the numbers assigned on an arbitrary basis. Rectangles are color-coded according to season: 1997–1998, orange; 1998–1999, yellow; 1999–2000, light green; 2001–2002, dark green; 2002–2003, light blue; 2003–2004, dark blue; and 2004–2005, purple; globally sampled background isolates are in red. The 2003–2004 clade V corresponds to the reassortant “clade B” defined previously [13]. Bootstrap values (>70%) are shown for key nodes. The tree is midpoint rooted for purposes of clarity, and all horizontal branch lengths are drawn to scale.
Figure 2. Phylogenetic Relationships of the HA Gene of Influenza Viruses Sampled from New York State and Globally during 1997–2005, Estimated Using an ML Method.

Rectangles represent clusters of related New York State isolates, with the size of the rectangle reflecting the number of isolates in the clade. Roman numerals indicate separate clades, with seasons colored as in Figure 1. Note that Roman numerals only denote the number of separate entries and do not correspond to the clade numbers given in Figure 1. Bootstrap values (>70%) are shown for key nodes. The tree is midpoint rooted for purposes of clarity, and all horizontal branch lengths are drawn to scale.
Viral Immigration Is a Major Contributor to Within-Season Genetic Diversity
For every season except for 2004–2005 (for which few global background isolates were available), the New York viruses do not form a monophyletic group. Rather, these isolates are interspersed with those sampled from other localities, often with strong bootstrap support. This pattern is indicative of widespread global gene flow into and out from New York State, rather than in situ evolution. For example, viral isolates that circulated during the same season in Greece, Argentina, Australia, and several other U.S. states fall within a single cluster of New York 1997–1998 isolates (clade III) on the NA tree (Figure 1). In addition, we observed little spatial structure within New York State, as individual clades contained viruses that were mixed by New York county of origin, indicative of active intrastate viral traffic.
We estimated the number of separate viral introductions into New York State as the number of phylogenetically distinct clades present in each season, which we identified as those topologically separated by viruses sampled in other localities and clades from other seasons, as well as by particularly long branch lengths. We considered the NA tree to be the more reliable estimator of the number of migration events since it usually contained more clades within each season than the HA tree, in which a single clade tends to predominate during each season. The existence of fewer HA clades most likely reflects a lack of background isolates in HA compared to NA, the fact that HA undergoes more frequent reassortment, and/or the action of periodic immune selection.
The NA phylogeny shows at least three separate introductions of viral isolates into New York State during the 1997–1998 season, six in 1998–1999, four in 1999–2000, three in 2001–2002, five in 2002–2003, five in 2003–2004, and one during the 2004–2005 season (Figure 1). These estimates are likely too conservative due to a paucity of background sequences, with particular underrepresentation of global viruses from low-frequency clades and from asymptomatic patients. Indeed, individual clades contain small groups of sequences defined by high bootstrap values, including four clusters with >70% bootstrap support within clade I from 2004–2005, which may represent additional introduction events. Separate entries may also be phylogenetically indistinguishable. For example, the three closely related clusters of sequences that comprise the 1997–1998 clade III could emerge as three separate introductions upon further global sampling.
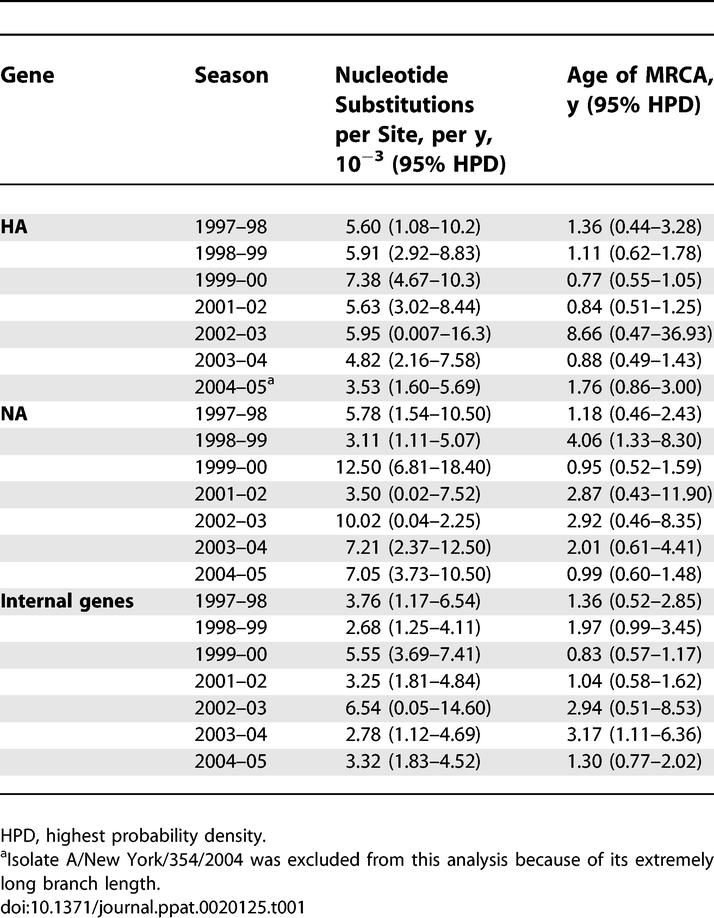
To further explore the genesis of intraseasonal genetic diversity, we estimated rates of nucleotide substitution and the age of the most recent common ancestor (MRCA) for the HA, NA, and concatenated internal genes (Table 1). Substitution rates estimated using a relaxed molecular clock to account for lineage-specific rate variation fell within the range typical of RNA viruses [16]: 3.53–7.38, 3.11–12.50, and 2.78–6.54 × 10−3 nucleotide substitutions per site, per year for the HA, NA, and concatenated internal genes, respectively. Mean MRCA ages ranged from 0.77 to 8.66 y for the HA gene, from 0.95 to 4.06 y for the NA gene, and from 0.83 to 3.17 y for the concatenated internal genes (Table 1). Given that each influenza season lasted from 15 to 28 wk, such relatively old MRCA values reveal that the ancestors of the influenza virus population in any season existed several months—or even years—prior to the start of that season, further indicating that isolates had already diversified before being imported into New York.
Table 1.
Nucleotide Substitution Rates and Ages of MRCA by Gene and Season
Viral Clades Observed in One Season Do Not Seed Those Observed in Later Seasons
If a single lineage dominated viral evolution, then we might expect the most successful viruses in a particular season to directly beget the next season's viral population through in situ evolution. Instead, the topological distance between clades on our phylogenetic trees shows that the H3N2 viruses circulating in New York State in a given season tend to derive from newly imported genetic material rather than from isolates circulating in New York State the previous season. A small number of clades may derive in situ from the past season's New York State viral population, including clades 1999–2000 IV, 1999–2000 I, 2002–2003 I, 2002–2003 II, and 2003–2003 I on the NA tree, as well as 1998–1999 VI on the HA tree. However, these always represent either individual viral isolates or minor clades with few progeny viruses, suggesting that even if in situ evolution has occurred between seasons, it has been a relatively minor contributor to overall genetic diversity. Furthermore, the limited sampling of global background strains may give the false appearance of in situ evolution.
Adaptive Evolution in HA Is Infrequent Within Seasons
Among the 22 total clades on the HA phylogeny observed over the seven seasons studied, amino acid differences between cocirculating clades were seen at 61 residues (Table 2). Of these, 41 fell at one of 131 amino acid positions located at or near the five antigenic regions of the HA1 domain [17–18]. While 14 changes were located at 18 antigenic sites previously proposed to experience strong positive selection [1], in all but three seasons this amounted to just a single amino acid difference. Although the 2002–2003 season exhibited the greatest number of amino acid differences among clades (22), isolates from the 1997–1998 season possessed the most changes at antigenic sites (14) and past positively selected sites (6), which corresponds to the Wuhan-to-Sydney antigenic transition [12]. At the intraclade level, 13 of the 22 clades exhibited amino acid variation (Table 2), comprising 80 amino differences at antigenic sites and nine at past selected sites. However, most variability again comprised only singleton mutations (41/80 changes at antigenic sites) in the largest clades.
Table 2.
Amino Acid Variation in the HA Gene of H3N2 Influenza A Virus from New York State 1997–2005

Most striking, however, was the overall weak effect of positive selection. For the New York State isolates sampled from all seasons, we examined the site-specific numbers of nonsynonymous (dN) versus synonymous (dS) substitutions, with dN > dS indicative of position selection. For those clades cocirculating within seasons, only two amino sites in HA (sites 13 and 236) displayed evidence of adaptive evolution (Table 2). Similarly, among a sample of viruses representing all clades from the entire study period we found evidence for positive selection only at site 160. None of these three sites have previously been shown to experience positive selection [1]. The lack of both positive selection and abundant variation observed at sites previously proposed to experience adaptive evolution suggests that most clades cocirculating within a season do not differ substantially in antigenicity and that immune escape mutations do not occur regularly over the course of a season. Further, with knowledge of the day upon which a virus was sampled (data provided in Table S1), we deduced that intraseasonal clades cocirculated contemporaneously, rather than displacing one another over the course of an epidemic season, as would have been expected had selection been acting upon major differences in fitness.
Interclade Reassortment Is Frequent
Viral isolates occupying incongruent positions on the HA, NA, and internal concatenated gene phylogenies provide evidence of hybrid genomes arising from reassortment. Overall, we detected 14 clear reassortment events involving 11 influenza isolates from four different seasons, several of which have been observed previously [13], thereby confirming the importance of this process in shaping the genetic diversity of influenza A virus (Table 3). Of these, five acquired a new HA gene, two a new NA gene, and three both new HA and NA genes through double reassortment, while a single isolate acquired one or more of the six internal genes. Of the new NA and HA genes acquired, six were positioned within a season's major or minor clade, while the remaining seven involved singletons. Notably, multiple clades on the NA and internal gene trees tended to combine into a single clade on the HA tree (such as A/New York/332/1999), suggesting that longer-term antigenic drift may favor these HA clades.
Table 3.
Reassortment Events in H3N2 Influenza A Virus from New York State, 1997–2005

Discussion
Although antigenic drift has been the dominant model for understanding epidemic dynamics, the full picture of influenza A virus evolution is more complex, including punctuated antigenic change [12], frequent reassortment [13–15], multiple cocirculating lineages [13], and little antigenic change over an epidemic season [19]. Hence, viral phylogenies based on HA1 in isolation and constructed from a relatively unrepresentative sampling of global isolates obscure some of the complex dynamics underlying influenza evolution in the short term and within discrete populations. Most notably, such phylogenies may fail to capture the full extent of viral genetic diversity and the processes of clade introduction, cocirculation, and reassortment, as well as the emergence of minor clades that could become dominant in later years.
Our study reveals that the genetic diversity of H3N2 influenza A virus during a single season is often extensive and to a large extent generated by independent introductions of genetically distinct viral isolates. No major clade of viruses in a given season appears to have evolved in situ from those that circulated locally in the prior season, indicating that the genetic diversity of influenza A virus in New York State is mostly replenished each season from an extensive global gene pool. This inference is further supported by the observation that viral populations in a given season have common ancestors that date back months or even years before the start of that season. While extensive geographic movement is therefore commonplace in areas such as New York State, whether this applies to smaller and more isolated populations needs to be investigated.
In addition, reassortment between clades occurs frequently, enabling genetically divergent isolates, which may be remnants from past seasons and other localities, to acquire new genetic material, including that which is selectively advantageous. Such frequent reassortment means that coinfection of individual hosts with multiple distinct viral clades must also be commonplace.
Of particular note is that adaptive evolution is infrequent within individual influenza seasons. The lack of positive selection within and among cocirculating clades suggests that most do not differ substantially in either antigenicity or fitness. This again supports the notion that the genetic diversity observed within seasons largely arises from the chance introduction of divergent isolates, rather than from immune selection operating over the course of a season. Hence, stochastic processes are more important in influenza virus evolution than previously thought, generating substantial genetic diversity in the short term. Such stochasticity will evidently impede attempts to predict the future course of viral evolution that assume influenza A virus evolution is largely a deterministic process that can be predicted by HA1 sequence analysis alone, and highlights the need for phenotypic data such as provided by “antigenic maps” [12]. Thus, antigenic drift appears to be a more sporadic process than previously thought, which raises the question of when most antigenic drift occurs: perhaps during local population bottlenecks, mainly coinciding with antigenic cluster jumps, or only under particular epidemiological conditions.
Broader and more intensive surveillance [20], combined with similar genomic analyses of additional populations, is fundamental to obtain a more comprehensive picture of global influenza diversity and to elucidate patterns and rates of global migration and reassortment [21]. In particular, the winter seasonality of human influenza A virus remains poorly understood, including the transmission routes by which viruses travel between Northern and Southern hemispheres and the importance of tropical regions as a possible reservoir of infection [22,23]. Tracking how clades circulate between Northern and Southern hemispheres using appropriate phylogenetic methods and robust global databases could further clarify what drives such seasonal patterning. Such studies may also enable the earlier identification of emerging dominant viral lineages and thereby assist vaccine strain selection.
Materials and Methods
Influenza viruses used in this study.
Viruses were collected by The Virus Reference and Surveillance Laboratory at the Wadsworth Center, New York State Department of Health, as part of the National Institute of Allergy and Infectious Disease's Influenza Genome Sequencing Project (http://www.niaid.nih.gov/dmid/genomes/mscs/influenza.htm) [8]. Samples were minimally passaged in primary rhesus monkey kidney cell culture, and RNA was extracted from the clarified supernatant. The Institute for Genomic Research derived whole-genome sequence information using methods described previously [8]. Accordingly, 413 complete genomes of influenza A virus (H3N2) sampled from New York State during the period 1997–2005 (excluding 2000–2001, for which only a single H3N2 sequence was available in an H1N1-dominant season) were downloaded from the National Center for Biotechnology Information (NCBI) Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Viruses were collected from all 11 regions within New York State with sampling dates available on GenBank (Table S1).
Sequence analysis.
Sequence alignments were manually constructed for the major coding regions of each segment, focusing on the HA (1,698 bp) and NA (1,407 bp). An alignment of the concatenated six internal gene segments was also constructed (9,636 bp), as these are expected to exhibit evolutionary patterns different from HA and NA. To place the New York State viruses in a global context, 48 unique HA gene sequences and 140 unique NA gene sequences from other human and swine influenza A viruses sampled worldwide from 1997–2005 were compiled from GenBank to make total datasets of 466 and 553 sequences for the HA and NA, respectively (Tables S2 and S3).
Phylogenetic trees were inferred for the HA, NA, and concatenated internal genes using the maximum likelihood (ML) method available in PAUP* [24]. In each case the Hasegawa-Kishino-Yano (HKY) 85 + I + Γ4 model of nucleotide substitution was employed, with the transition–transversion ratio, proportion of invariant sites (I), and the gamma distribution of among-site rate variation with four rate categories (Γ4) estimated from the empirical data (parameter values available from the authors on request). Because of the very large size of all datasets, the nearest-neighbor-interchange branch-swapping method was employed. To assess the robustness of individual nodes on the phylogenetic tree, we performed a bootstrap resampling analysis (1,000 replications) using the neighbor-joining procedure but incorporating the ML substitution model. Independent entries of A(H3N2) viruses into New York State were determined by identifying viral isolates that were separated from the others circulating in that season (1) by viruses sampled from localities outside of New York State, and (2) by exceptionally long branch lengths.
Rates of nucleotide substitution and age of the MRCA were estimated using a Bayesian Markov Chain Monte Carlo (MCMC) method available in the BEAST package [25], which considers the distribution of branch lengths among viruses sampled at different times (day of sampling). Uncertainty in the data is reflected in the 95% highest probability density values. This analysis employed the HKY85 substitution model assuming exponential population growth and a relaxed (uncorrelated exponential) molecular clock which consistently best fit the data. In all cases, chains were run until convergence was achieved.
We used the MacClade program [26] to determine those amino acid changes in the HA gene that occurred within and among each clade, particularly those at (1) 131 amino acid positions in five antigenic regions of the HA1 domain [17–18], and (2) 18 antigenic sites in the HA1 domain that have previously been proposed to experience positive selection [1]. Site-specific selection pressures in HA (New York State viruses alone) were measured as the ratio of dN to dS per site estimated using the single likelihood ancestor counting (SLAC; all sequences per season) and fixed effects likelihood (FEL; maximum of 50 randomly sampled sequences per season) methods, both incorporating the general reversible substitution (REV) model with phylogenetic trees inferred using the neighbor-joining method available at the Datamonkey facility [27]. This analysis was undertaken on the intraseasonal data and an interseason dataset comprising a random sample of three isolates from each clade and all singletons (n = 52 isolates).
Supporting Information
Rectangles represent clusters of related New York isolates, with the size of the rectangle reflecting the number of isolates in the clade. Roman numerals indicate separate clades, with seasons colored as in Figure 1. Roman numerals denote the number of separate entries and do not correspond to clade numbers given in Figure 1. Bootstrap values (>70%) are shown for key nodes. The tree is midpoint rooted for purposes of clarity, and all horizontal branch lengths are drawn to scale.
(769 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(136 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(111 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(72 KB PDF)
The GenBank accession numbers shown are for the PB2 gene only. Accession numbers for the remaining segments of each viral isolate are available at NCBI's Influenza Virus Resource: http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/select.cgi.
(816 KB DOC)
(97 KB DOC)
(49 KB DOC)
Accession Numbers
Accession numbers for the remaining segments of each viral isolate in Table S2 are available at NCBI's Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/select.cgi).
Acknowledgments
Viruses described in this study collected after 2001 include some isolates collected as part of the Sentinel Physician Influenza Surveillance Program, supported by Cooperative Research Agreement Number U50/CCU223671 from the Centers for Disease Control and Prevention. Laboratory support was provided by M. Kleabonas and R. Bennett at the Wadsworth Center.
Abbreviations
- dN
nonsynonymous substitutions
- dS
synonymous substitutions
- HA
hemagglutinin
- ML
maximum likelihood
- MRCA
most recent common ancestor
- NA
neuraminidase
Footnotes
Competing interests. The authors have declared that no competing interests exist.
Author contributions. MIN and ECH conceived the study and wrote the paper, with contributions from all other authors. JT, KSG, and SGB collected the viral samples. EG, NAS, and DJS undertook the viral genome sequencing. LS, CV, MAM, and BTG advised on epidemiological aspects of the study. MIN, IV, and ECH performed the sequence analysis. DJL and JKT wrote the paper.
Funding. The authors received no specific funding for this study.
References
- Bush RM, Fitch WM, Bender CA, Cox NJ. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999;16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- Bush RM Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286:1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Ferguson NM, Galvani AP, Bush RM. Ecological and immunological determinants of influenza evolution. Nature. 2003;422:428–433. doi: 10.1038/nature01509. [DOI] [PubMed] [Google Scholar]
- Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Phil Trans R Soc Lond B. 2001;356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM, Leiter JME, Li X, Palese P. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci U S A. 1991;88:4270–4274. doi: 10.1073/pnas.88.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM, Bush RM, Bender CA, Cox NJ. Long term trends in the evolution of H(3) HA1 human influenza. Proc Natl Acad Sci U S A. 1997;94:7712–7718. doi: 10.1073/pnas.94.15.7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Dushoff J, Levin SA. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc Natl Acad Sci U S A. 2002;99:6263–6268. doi: 10.1073/pnas.082110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, Sengamalay NA, Shumway M, Zaborsky J, Feldblyum T, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- Bridges CB, Thompson WW, Meltzer MI, Reeve GR, Talamonti WJ, et al. Effectiveness and cost-benefit of influenza vaccination of healthy working adults: A randomized controlled trial. JAMA. 2000;284:1655–1663. doi: 10.1001/jama.284.13.1655. [DOI] [PubMed] [Google Scholar]
- de Jong JC, Beyer WEP, Palache AM, Rimmelzwaan GF, Osterhaus AD. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J Med Virol. 2000;61:94–99. [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Preliminary assessment of the effectiveness of the 2003–04 inactivated vaccine—Colorado, December 2003. Morb Mortal Wkly Rep. 2004;53:8–11. [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Holmes EC, Ghedin E, Miller N, Taylor J, Bao Y, et al. Whole genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 2005. e300. [DOI] [PMC free article] [PubMed]
- Lin YP, Gregory V, Bennett M, Hay A. Recent changes among human influenza viruses. Virus Res. 2004;103:47–52. doi: 10.1016/j.virusres.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Lindstrom SE, Cox NJ, Klimov A. Genetic analysis of human H2N2 and early H3N2 influenza viruses, 1957–1972: Evidence for genetic divergence and multiple reassortment events. Virology. 2004;328:101–119. doi: 10.1016/j.virol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Jenkins GM, Rambaut A, Pybus OG, Holmes EC. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic approach. J Mol Evol. 2002;54:156–165. doi: 10.1007/s00239-001-0064-3. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–379. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- Lavenu A, Leruez-Ville M, Chaix ML, Boelle PY, Rogez S, et al. Detailed analysis of the genetic evolution of influenza virus during the course of an epidemic. Epidemiol Infect. 2005;134:514–520. doi: 10.1017/S0950268805005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RMA, Kuiken T, Rimmelzwaan G, Osterhaus AD. Global task force for influenza. Nature. 2005;435:419–420. doi: 10.1038/435419a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ. Predictability and preparedness in influenza control. Science. 2006;312:392–394. doi: 10.1126/science.1122665. [DOI] [PubMed] [Google Scholar]
- Viboud C, Alonso WJ, Simonsen L. Influenza in tropical regions. PLoS Med. 2006. e89. [DOI] [PMC free article] [PubMed]
- Cox NJ, Subbarao K. Global epidemiology of influenza: Past and present. Annu Rev Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic analysis using parsimony (*and other methods), version 4.0 [computer program] Sunderland, (Massachusetts): Sinauer Associates; 2003. [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006. e88. [DOI] [PMC free article] [PubMed]
- Maddison DR, Maddison WP. MacClade. Analysis of Phylogeny and Character Evolution, version 4.0 [computer program] Sunderland, (Massachusetts): Sinauer Associates; 2000. [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rectangles represent clusters of related New York isolates, with the size of the rectangle reflecting the number of isolates in the clade. Roman numerals indicate separate clades, with seasons colored as in Figure 1. Roman numerals denote the number of separate entries and do not correspond to clade numbers given in Figure 1. Bootstrap values (>70%) are shown for key nodes. The tree is midpoint rooted for purposes of clarity, and all horizontal branch lengths are drawn to scale.
(769 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(136 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(111 KB PDF)
The tree is midpoint rooted for purposes of clarity and all horizontal branch lengths are drawn to scale.
(72 KB PDF)
The GenBank accession numbers shown are for the PB2 gene only. Accession numbers for the remaining segments of each viral isolate are available at NCBI's Influenza Virus Resource: http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/select.cgi.
(816 KB DOC)
(97 KB DOC)
(49 KB DOC)