

Abstract
Within the first 5 min after a sudden relief from glucose limitation, Saccharomyces cerevisiae exhibited fast changes of intracellular metabolite levels and a major transcriptional reprogramming. Integration of transcriptome and metabolome data revealed tight relationships between the changes at these two levels. Transcriptome as well as metabolite changes reflected a major investment in two processes: adaptation from fully respiratory to respiro-fermentative metabolism and preparation for growth acceleration. At the metabolite level, a severe drop of the AXP pools directly after glucose addition was not accompanied by any of the other three NXP. To counterbalance this loss, purine biosynthesis and salvage pathways were transcriptionally upregulated in a concerted manner, reflecting a sudden increase of the purine demand. The short-term dynamics of the transcriptome revealed a remarkably fast decrease in the average half-life of downregulated genes. This acceleration of mRNA decay can be interpreted both as an additional nucleotide salvage pathway and an additional level of glucose-induced regulation of gene expression.
Keywords: glucose pulse, metabolome, Saccharomyces cerevisiae, systems biology, transcriptome
Introduction
It is essential for any organism to rapidly and efficiently adjust its metabolism and physiology to changes in nutrient availability and other environmental parameters (Causton et al, 2001; Gasch and Werner-Washburne, 2002). In the yeast Saccharomyces cerevisiae, nutrient responses have been most extensively studied for glucose, the preferred carbon and energy source for this yeast (see for review Gancedo, 1998; Rolland et al, 2002). Changes in extracellular glucose availability trigger a variety of cellular responses.
Addition of glucose to S. cerevisiae cells that exhibit a fully respiratory metabolism elicits a range of transcriptional, translational and post-translational modifications. These changes are preceded and, to a large extent, triggered by changes of intracellular metabolites and low-molecular-weight effectors. Changes of intracellular metabolite pools occur within seconds of a perturbation of the extracellular glucose concentration. For example, after a glucose pulse to respiring cells, the concentrations of metabolites of the upper part of glycolysis (e.g. fructose-6-phosphate (F6P) and fructose-1,6-bisphosphate (F1,6P2)) rapidly increase, whereas those of metabolites from the lower part of glycolysis (e.g. 2- and 3-phosphoglycerate (2PG, 3PG) and phosphoenolpyruvate (PEP)) rapidly decrease (Theobald et al, 1993; Visser et al, 2004). These changes have a strong impact on the energy status of the cells. Immediately after a glucose pulse, intracellular ATP concentration decreases, whereas ADP and AMP levels slightly increase, thus leading to a decrease in the energy charge. Remarkably, a substantial decrease in the overall adenine nucleotide (‘AXP') pools is reproducibly observed in studies on the fast dynamics of glucose responses in S. cerevisiae (Theobald et al, 1997). This phenomenon is among the aspects of glucose responses in yeast that remain to be elucidated.
In addition to metabolites and cofactors, perturbation of the extracellular glucose concentration causes rapid changes of second messenger molecules such as cAMP (Thevelein et al, 2005) and D-myo-inositol-(1,4,5)-triphosphate (IP3) (Belde et al, 1993). These in turn contribute to responses at the transcriptional level and at the post-transcriptional level, where glucose triggers the specific inactivation and proteolysis of many proteins, including the gluconeogenic enzymes fructose-1,6-biphosphatase and several hexose transporters via a process called catabolite inactivation (Mazon et al, 1982; Mercado et al, 1991).
The most extensively documented way in which glucose affects transcription is called glucose catabolite repression and encompasses the coordinated downregulation of the transcription of large groups of genes involved in respiration, metabolism of non-glucose carbon sources and several hexose transporters (Gancedo, 1998). In addition to a downregulation of transcription, glucose induces accelerated degradation of specific mRNAs, such as the transcript of SDH1 and SDH2 that encode subunits of succinate dehydrogenase (Lombardo et al, 1992; Cereghino et al, 1995) and SUC2 that encodes invertase (sucrose utilization) (Cereghino and Scheffler, 1996).
For a quantitative systems analysis of the dynamic responses to glucose availability, it is essential that experimental conditions are tightly controlled. Steady-state chemostat cultures are excellently suited as a reproducible and stable experimental baseline (Hoskisson and Hobbs, 2005; Ronen and Botstein, 2006). A typical experimental design then consists in the application of a defined perturbation (e.g. a glucose pulse) to a steady-state chemostat culture, followed by rapid sampling, quenching of metabolism and analysis of relevant intracellular and extracellular components (Theobald et al, 1997).
So far, analysis of the rapid transient (timescale 1 s to 5 min after a perturbation) has mainly been studied at the metabolome level (Theobald et al, 1997; Visser et al, 2004). An often-implicit assumption in these studies is that, over these short time periods, the concentrations of active enzymes in the cells remain constant. In that case, the measured responses allow for direct identification and quantification of kinetic interactions at the metabolome level. However, verification of this important assumption by simultaneous analysis of gene expression at the transcriptional or translational level has so far not been attempted.
The present study represents the first dedicated attempt to integrate quantitative datasets obtained at the metabolite and transcript level during the first minutes after a defined metabolic perturbation of S. cerevisiae. To this end, we analyzed levels of key metabolites in primary metabolism as well as genome-wide mRNA levels in the first 5 min after glucose pulse to aerobic, glucose-limited chemostat cultures of yeast. To investigate the apparent lack of conservation of the adenine nucleotide pool observed in previous studies, special attention was paid to the dynamics of purine metabolism. Our results provide new insights into the chronology of events between the metabolic and the primary transcriptional responses to glucose in S. cerevisiae and show a biologically significant correlation between metabolome and transcriptome with respect to energy requirement and nucleotide metabolism during the initial phase of growth acceleration after glucose pulse.
Results and discussion
Global transcriptional responses following a glucose pulse
In glucose-limited cultures of S. cerevisiae where metabolism is fully respiratory, the very low residual glucose concentration (0.15 mM) was instantaneously increased to 5.6 mM by pulsing a concentrated glucose solution (Figure 1A). Three independent cultures were pulsed with glucose and samples for transcriptome analysis were taken at various time points up to 330 s after glucose addition. These three independent pulses were highly reproducible and the average coefficient of variation for transcript levels measured at replicate time points was below 19% (Supplementary information 1).
Figure 1.

Response of glucose-limited chemostat (D=0.05/h) to a 5.6 mM glucose pulse. (A) Extracellular concentration of glucose (•), ethanol (▪), glycerol (▾) and acetate (▴) are plotted as a function of time (s). Two independent pulse experiments are represented. (B) Two-dimensional clustering heat-map of the differentially expressed genes in the glucose pulse experiment. Each expression datum represents the average of at least two independent replicates. Orange (relatively high expression) and blue (relatively low expression) squares were used to represent the transcription profiles of genes deemed specifically changed. K-means clusters of genes with ascendant profiles (A, B and C) and descendent profiles (D, E). The thick black line represents the average of the median normalized expression data of the genes comprising the cluster.
Multiclass statistical analysis yielded a set of 1154 genes that displayed significant changes in transcription between at least two time points. Analysis of this set of genes by K-means clustering identified five glucose-responsive gene clusters (Figure 1B). Clusters A, B and C (589 genes) grouped genes the expression of which was increased after glucose addition, whereas clusters D and E (565 genes) showed the opposite trend (Figure 1B) (Supplementary information 2). Significant changes in genes transcription only started between 120 and 210 s after the glucose pulse (Figure 1), thus providing an exact quantification of the time required for glucose signal transduction and activation of transcription.
Glucose-responsive transcripts were subsequently analyzed to assess the enrichment of functional categories (Figure 2A and Supplementary information 3). The gene clusters that were transcriptionally upregulated after the glucose pulse showed a significant enrichment of metabolic functions and more specifically of amino-acid, purine ribonucleotide and nucleotide metabolism. Other significantly overrepresented categories among the upregulated transcripts were involved in the transcription, synthesis and processing of ribosomal RNA (Figure 2A). The gene clusters downregulated after the glucose pulse exhibited a significant enrichment in the ‘energy and metabolism' functional categories (Figure 2A). This global analysis revealed that drastic metabolic rearrangements are set in motion in the first minutes after release from glucose limitation.
Figure 2.

Interpretation of transcriptome data. (A) The 1154 differentially expressed genes were distributed over MIPS functional categories as a function of time (s). The number mentioned in brackets indicates the total number of genes found in the categories. Overrepresented primary and secondary functional categories according to a hypergeometric distribution analysis with a threshold P-value of 0.01 with Bonferroni correction are mentioned together with their calculated P-values. (B) Transcription factor analysis; the 1154 differentially expressed genes were intersected with transcription factor target genes according to the ChIP on chip analysis (Harbison et al, 2004) and the probability that the representation of each factor occurred by chance was assessed by hypergeometric distribution. The table displays significant factors that returned a P-value lower than 0.05.
In order to identify the regulatory networks responsible for the transcriptional response to the glucose pulse, our dataset was combined with the genome-wide yeast location analysis datasets for 102 transcription factors from Harbison et al (2004). Thus, 12 transcription factors could be assigned to the clusters of upregulated genes with high confidence (Figure 2B) (Supplementary information 4). The function of these transcription factors obviously overlapped with the enriched functional categories. The most overrepresented factor was Bas1p, which corroborated the enrichment in purines and nucleotides metabolism categories. In addition to Bas1p, several sets of transcription factors could be distinguished and assigned to specific cellular functions (Figure 2B). A first coherent set including Met4p, Met31p, Met32p and Cbf1p, all members of a transcriptional complex, revealed a major transcriptional reprogramming of sulfur metabolism (Rouillon et al, 2000). Gcn4p and Leu3p are involved in amino-acid metabolism and biosynthesis. Fhl1p, Rap1p and Abf1p could be intuitively connected to ribosome biogenesis transcriptional control (Lascaris et al, 2000; Martin et al, 2004; Rudra et al, 2005). However, the involvement of Ash1p (involved in filamentous growth; Pan and Heitman, 2000) and Swi4p (cell cycle; Nasmyth and Dirick, 1991) could not be predicted from the enriched functional categories (Figure 2A).
The 12 transcription factors found significantly linked to the clusters of downregulated genes were in good agreement with the transcriptional network involved in glucose catabolite repression (Figure 2B), such as the Cyc8p-Tup1p-associated factors Nrg1p and Sko1p, and general regulator such as Ume6p (Williams et al, 2002) and the activator of the gluconeogenic regulon Sip4 (Schuller, 2003), known to be repressed in the presence of excess glucose. Additionally, overrepresentation of Msn2p and Msn4p, STRE (stress responsive element) transcription factors, which are part of Gpr1p/Gpa2p glucose-sensing pathway (Gelade et al, 2003), was observed, completing this regulatory network.
Addition of glucose to carbon-limited chemostat cultures results in a drain of the adenine nucleotides
The 5.6-mM glucose pulse to aerobic, carbon-limited cultures resulted in an immediate increase in the rate of glucose consumption. As described previously, the acceleration of glucose consumption was accompanied by switching to respiro-fermentative metabolism (Visser et al, 2004), evidenced by the accumulation of ethanol and, to a lesser extent, acetate and pyruvate in the cultures (Figure 1A). Intracellular metabolites were analyzed with a particular emphasis on mono-, di- and triphosphate nucleotides (NXP). As previously shown (Theobald et al, 1993, 1997), an immediate dramatic decrease of intracellular ATP concentration and a concomitant increase in AMP were observed, followed by slow recovery (Figure 3). However, contrary to earlier assumption, this drop in ATP could neither be entirely attributed to the hydrolysis of ATP for energy transfer process such as glucose phosphorylation nor to the increase in RNA synthesis (Theobald et al, 1997). First of all, the net increase in AMP and ADP did not balance the ATP loss. The adenine moiety pool (ATP, ADP plus AMP) was not conserved over time: after a clear drop within the first 60 s, the sum AXP rose (Figure 3). Secondly, the profiles of the UXP, CXP and GXP showed similar initial decreases compared to the AXP profiles, albeit in different absolute level, and the amplitude of the GXP drop was 20-fold lower than for the AXP pool (Figure 3). The U, G and C nucleotide pools returned to their initial concentrations or increased beyond those within the first 200 s after the glucose pulse (see Supplementary information 5) (Figure 3F and G). As RNA biosynthesis consumes all four nucleotides (ATP, UTP, GTP and CTP) in a 0.254:0.246:0.226:0.274 molar ratio (Herbert et al, 1971), AXP consumption for RNA biosynthesis can be calculated from the lowest drop in nucleotide pools, that is, CXP, assuming that the biosynthesis of the nucleotides does not immediately increase to the demand. This calculation reveals that RNA biosynthesis would only account for 5% of the decreased AXP pool (Figure 3C). Estimation of the other ATP-consuming processes compatible with an increase in the growth rate to its maximum (CEN PK 113-7Dμmax=0.45/h; van Dijken et al, 2000) in DNA, histidine and cofactor biosynthesis is far from sufficient to explain the observed drop in sum AXP (Figure 3I and J and Supplementary information 6). This clearly indicated that the AXP nucleotide pool was involved in unknown processes during the first 60 s after the addition of glucose. Quantitative determination of other possible adenine moiety sinks, free adenosine, adenine, hypoxanthine, nicotinamide adenine dinucleotide (NAD/NADH) and other adenosine-containing molecules such as S-adenosylmethionine, S-adenosylhomocysteine, or even activated sugars (ADP- and UDP-glucose), is of primary importance to understand this still unsolved phenomenon. The LC-MS/MS methods for the quantification of these metabolites are still under development.
Figure 3.

Intracellular concentrations of mono-, di- and triphosphate nucleotides (AXP, CXP, GXP, UXP) in micromoles per gram of dry weight (μmol/g DW) following 5.6 mM glucose pulse. (A) ATP, (B) ADP, (C) AMP, (D) e-charge defined as the ratio (ATP+0.5 ADP)/(ATP+ADP+AMP), (E) ΣAXP, (F) ΣGXP, (G) ΣCXP, (H) ΣUXP and (I) possible adenine nucleotide utilization. ATP, ADP, AMP and cAMP comprise the adenine nucleotide (AXP) pool in which any reaction between them does not cause any depletion in the pool size. Outside the circle are the reactions that are consuming the adenine moiety of the AXP. Adapted from Chapman and Atkinson (1977). (J) Theoretical distribution of AXP based on change in the synthesis rate owing to growth acceleration (from μ=0.05/h to μmax=0.45/h). Detailed calculations are provided in Supplementary information 6. The data plotted originate from at least two independent pulse experiments.
Following its early drop, the AXP pool recovered at a rate of approximately 0.01 μmol/g DW/s (calculated from the total nucleotide pool slope), whereas at steady state the net adenine nucleotide synthesis rate was only 0.0001 μmol /g DW/s (calculated from AXP concentration at steady state at a growth rate of 0.05/h; see Supplementary information 6), that is, about two orders of magnitude lower than the observed recovery rate. This implies a strong increase in the adenine biosynthesis rate and an important role of the salvage pathway.
Metabolic inter-relations explain transcriptome co-responses: the adenine nucleotide pool drain is accompanied by upregulation of purine biosynthesis, C1 and sulfur metabolism
Consistent with the drop in adenosine nucleotide pool that has been previously discussed, the genes of the de novo purine biosynthesis pathway, by which the AXP pool is synthesized, were found significantly overrepresented among the upregulated genes (Figure 2A). All but one of the 13 genes composing that pathway were upregulated (Figure 4); only ADE16, encoding a bifunctional IMP cyclohydrolase—phosphoribosyl-amino imidazole carboxamide formyltransferase, was expressed constitutively. The expression of genes encoding one-carbon (C1) metabolism such as SHM2, MTD1 and ADE3 was concurrently upregulated (Figure 4). In addition to purine biosynthesis, the C1 metabolism, using folate coenzymes, is essential for glycine, methionine and methyl group biogenesis. Genes encoding mitochondrial glycine cleavage pathway (GCV1, GCV2, GCV3), genes encoding methionine biosynthesis (MET3, MET14, MET16, MET28, MET31, MET32, MET2 and MET6) and S-adenosylmethionine (methyl donor) biosynthesis (SAM1 and SAM2) were also upregulated accordingly (Figure 4). Piper et al (2000) proposed a model in which cytosolic 5,10 methylene-THF is mainly directed to methionine biosynthesis for methylation reaction and mitochondrial one-carbon units derived from glycine are directed to purine biosynthesis. The simultaneous upregulation of the GCV genes encoding mitochondrial glycine decarboxylase and SHM2 encoding the cytosolic serine-hydroxymethyltransferase clearly suggested that not only purine but also generation of S-adenosylmethionine was important for the cell upon glucose exposure. Furthermore, the co-regulation of both folate metabolism branches revealed the fast utilization and recycling of 5,10 methylene-THF, as the upregulation of the GCV genes is an indicator of a low level of 5,10 methylene-THF (Piper et al, 2000). To further support the hypothesis that C1 metabolism is central in the transition described here, the genes encoding serine biosynthesis pathway (SER1, SER2, SER3 and SER33), which converts 3-phosphoglycerate to serine, were all significantly upregulated. Serine is indeed a co-substrate with THF for glycine and 10-formyl THF biosynthesis (Figure 4).
Figure 4.

Coordinated upregulation of the purine biosynthesis, sulfur assimilation and methionine and adenine salvage pathways. Metabolic pathways: The numbers indicated represent the fold change calculated between the expression values obtained at 330 s and the values obtained at the initial steady state (0 s).
The transcriptional regulation of the purine biosynthesis and part of the 10-formyl THF (SHM2 and MTD1) pathways has been shown to be under the control of Bas1p, a myb-like transcriptional activator (Denis et al, 1998; Denis and Daignan-Fornier, 1998). In agreement with this, the transcript level of BAS1 itself was coordinately upregulated more than two-fold. Integration of the data presented in this study and the supporting Bas1p location analysis by chromatin immunoprecipitation data (Harbison et al, 2004) agreed on the regulation of the glycine cleavage pathway (ADE3, GCV1, GCV2 and GCV3) by Bas1p as well. These results were also supported by the presence of TGACTC Bas1p binding site in the promoter of the latter genes. Altogether, these data would confirm the regulation by Bas1p of both purine and C1 metabolism derived from glycine.
On the other hand, a complex including differentially expressed MET28, MET31 and MET32 transcriptionally regulated the sulfur metabolism in a time-dependent manner (Figure 2). Genes encoding methionine uptake transport system (MUP1, MUP3), sulfate assimilation to methionine (MET3, MET14, MET16, MET2, MET6) and formation of methyl donor AdoMet (SAM1, SAM2) were all upregulated. Adomet, a sulfur-containing compound that functions as a methylating agent, may reflect an increase in methylation processes, as will be discussed in the following section.
Finally, the methyl transfer converts Adomet to S-adenosylhomocysteine, which can be recycled to methionine via a few steps, in which an adenosine moiety is released. The gene involved in this pathway, SAH1, was also found to be significantly upregulated (Figure 4A). The released adenosine can be recycled to the adenosine nucleotide pool via the purine salvage pathway (involving upregulated AAH1, HTP1) reducing the cost of AMP synthesis via de novo purine biosynthesis pathway (which requires five ATP and one GTP to form one AMP molecule from PRPP).
Although the metabolic crosstalks are quite apparent from a biochemical network, the regulatory network that coordinates the upregulation of genes involved in de novo purine biosynthesis, serine biosynthesis, THF metabolism, sulfur metabolism and purine salvage pathway is not trivial. Alternately, upregulation of purine and THF metabolism on the one hand and sulfur metabolism on the other can be explained as discussed above. However, no available reports can relate serine biosynthesis gene regulation to THF metabolism, as current transcriptome and metabolome data seem to relate them to one another.
Although some phenotypic evidences relate the serine biosynthetic pathway to purine metabolism, as a mutation in SER1 (initially named ADE9) leads to an adenine requirement, no molecular basis had been demonstrated so far (Buc and Rolfes, 1999). While the search for BAS1 binding motif (TGACTC) in the promoter sequences of the SER1, SER2, SER3 and SER33 identified this binding motif in SER2 and SER33, location analysis data for BAS1 failed to report any binding activity on SER gene promoters. However, ChIP on chip data revealed that the SER33 promoter sequence was bound by Cbf1 (Harbison et al, 2004), member of the Cbf1/Met30/Met4/Met28 complex (Thomas and Surdin-Kerjan, 1997; Blaiseau and Thomas, 1998) that regulates sulfur metabolism. Furthermore, genome-wide transcriptome analysis of S. cerevisiae grown in chemostat revealed that SER33 was specifically upregulated under sulfur limitation (Tai et al, 2005). These experimental facts suggest that cytosolic processes leading to C1 transfer for methionine and Adomet biosynthesis (serine biosynthesis, 5-methyl-THF synthesis) are coordinately controlled by central sulfur metabolism regulation.
Ribosome biogenesis is upregulated after relief from glucose limitation
The higher requirement of methylation substrate as deduced from the data mentioned above could be sustained as many genes involved in ribosomal RNA synthesis, processing and modification were upregulated following glucose addition (Figure 2A). This major induction of rRNA synthesis and ribosome biogenesis is indicative of a rapid synthesis and recruitment of the translational machinery. Among the 565 genes displaying a continuous increase in expression after pulsing glucose, 180 were related to transcription and protein synthesis of which 145 were involved in the assembly and activity of the translation machinery (Figure 5). Although the microarrays used in this study cannot provide quantitative information on rRNA, the upregulation of the components of the machinery involved in their transcription suggested an increased transcription of the genes encoding for rRNA. Indeed five subunits (RPA12, RPA135, RPA34, RPA43, RPA49) and two essential initiation factors (RRN7 and RRN11) of the RNA polymerase I (RNA-pol I) involved in the transcription of the rDNA were upregulated. Four additional genes (RPB10, RPB8, RPB5, RPO26) encoding subunits shared by RNA-pol I, II and III and RPC19 encoding a shared subunit of RNA-pol I and III were also upregulated. Besides, seven genes encoding subunits of either RNA-pol II (RPB9, RPB11, ROX3) or RNA-pol III (RPC11, RPC31, RPC37, RPC82) displayed increasing transcription profiles. These data clearly illustrated the concerted upregulation of all three RNA polymerases. In conjunction with an upregulation of the RNA-pol I subunits, 23 genes coding for ribosomal proteins and 121 genes encoding proteins involved in processing, maturation, export, modification and transcription of rRNA and ribosome components shared a similar increase in expression (Figure 5).
Figure 5.

Genes upregulated during the glucose pulse involved in transcription and translation functions according to MIPS categories. The independent replicate transcriptome datasetss for each time point were averaged and then compared. Blue (relatively low expression) and orange (relatively high expression) squares are used to represent the transcription profiles of genes deemed specifically changed.
The ribosomes undergo modifications such as conversion of uridines into pseudouridines and addition of methyl group to specific nucleotides with a majority at the 2′-O position of the ribose (Bachellerie and Cavaille, 1997). Consistently, five genes participating in Adomet-dependent methylation activity were upregulated (NOP1 +2.1, NOP58 +2.7, SNU13 +2.9, SPB1 +2.3, DIM1 +2.0). In good agreement with literature, FHL1 and RAP1 (transcription factors involved in transcriptional control of ribosome biogenesis) targets were significantly overrepresented within the set of upregulated genes (Figure 2B). The significant upregulation of genes encoding specialized methyltransferases involved in translation initiation (GCD10 +2.0 and GCD14 +1.9) and tRNA modifications (NCL1+2.3, TRM82 +2.0, TRM2 +2.1, TRM7 +1.8) indicated the importance of Adomet role in the metabolic circumstance described in this study (Figure 5).
The role of methylation reactions using Adomet should be taken into consideration in explaining a part of the drain of the AXP pools in the first minute following the addition of glucose (Figure 3). As shown here, this hypothesis would be in line with the upregulation of the purine and the methionine salvage pathways in response to the increase of S-adenosylhomocysteine when Adomet is used as methyl donor.
New insight into central carbon metabolism by integration of metabolite and transcript levels
The transcriptome analysis of the response of S. cerevisiae to a sudden relief from glucose limitation classified 565 genes with downregulated transcription (clusters D and E; Figure 1B). These clusters showed a specific enrichment for genes involved in energy generation and metabolism (Figure 2A). In previous chemostat-based studies (Boer et al, 2003; Tai et al, 2005), 19 genes exhibited consistent repression at high glucose concentration, irrespective of the limiting macronutrient (nitrogen, sulfur or phosphorus). In the current study, which applied dynamic glucose perturbation, 15 of these genes were found downregulated (JEN1, CSR2, HXK1, SUC2, SUC4, ISF1, GAL4, SOL1, MRK1, YLR327C, YFR017C, YER067W, YGR243W, YIL057C, YMR206W) confirming the occurrence of glucose repression even within the short time interval of 330 s.
In addition, the integration of the central carbon metabolism metabolite data with transcript analysis allows better understanding of the very early metabolic response of the cell facing a sudden increase of environmental glucose concentration. As previously reported (Visser et al, 2004), a rapid and transient increase of the metabolites of the top part of the glycolysis (Figure 6A–C) was observed, whereas the metabolites of the lower part followed the opposite trend (Figure 6E and F) (Theobald et al, 1993). This metabolite distribution was regarded as a direct consequence of the rate-limiting phosphofructokinase activity (Theobald et al, 1997). However, the constant increase of the F1,6-P2/F6P concentration ratio (as calculated from Figure 6B and C) contradicts this initial hypothesis and instead supports the hypothesis that the increase of the glyceraldehyde-3-phosphate dehydrogenase reaction rate and the delayed increase in ethanol formation (Figure 1A) affect the redox status of the cell, as shown by the large increase of NADH/NAD ratio (Figure 6O). This increase likely inhibits glyceraldehyde-3-phosphate dehydrogenase, which explains the observed reduction of metabolite concentrations of lower part of the glycolysis (Figure 6E and F).
Figure 6.

Intracellular concentration of glycolytic and TCA cycle intermediates following 5.6 mM glucose pulse, expressed in micromoles per gram of dry weight (μmol/g DW) except mentioned otherwise. (A) Glucose-6-phosphate (G6P), (B) fructose-6-phosphate (F6P), (C) fructose-1,6-biphosphate (F1,6P2), (D) fructose-2,6-biphosphate (F2,6P2) expressed as normalized to the steady-state value (see Materials and methods), (E) 2 and 3 phosphoglycerate (2PG+3PG), (F) phosphoenolpyruvate (PEP), (G) 6-phosphogluconate (6PG), (H) glucose-1-phosphate (G1P), (I) trehalose-6-phosphate (T6P), (J) citrate (CIT), (K) α-keto-glutarate (αKG), (L) succinate (SUC), (M) fumarate (FUM), (N) malate (MAL) and (O) NADH/NAD ratio expressed as normalized to the steady-state value. The data plotted originate from at least two independent pulse experiments.
To restore redox homeostasis, yeast produces ethanol and glycerol (Figure 1A) and fine-tunes the tricarboxylic acid (TCA) cycle, which is a source of reduced cofactor. In contrast to regulation of glycolysis in steady-state cultures, which predominantly takes place at the post-transcriptional level (Daran-Lapujade et al, 2004), TCA cycle regulation was visible at metabolome and transcriptome levels. The concentration of TCA cycle intermediates such as malate, fumarate and α-ketoglutarate increased to reach a new pseudo-steady-state level, whereas the citrate concentration was constant throughout the pulse experiment (Figure 6J–N), which indicates flux discontinuation from α-ketoglutarate to C4 pool (metabolite concentrations are provided in Supplementary information 7). This complies with the previous observation that under respiro-fermentative condition, the TCA cycle is not performing as a cycle but as two separate branches: an oxidative branch from pyruvate to α-ketoglutarate and a reductive branch from pyruvate to malate and fumarate (Gombert et al, 2001). Moreover, the transcriptome data clearly illustrate rapid transcriptional responses of the structural genes encoding TCA cycle enzymes (Figure 7). Eleven genes (KGD2, MDH1, SDH3, SDH1, ACO1, IDP3, MDH2, IDH2, LSC1, YMR118C, YLR164W) involved in the TCA cycle were immediately downregulated, whereas CIT2 and PYC1 were upregulated (Figure 7A). Transcription of HAP4, which encodes the activator of the Hap2p/3p/4p/5p complex involved in the transcriptional regulation of TCA cycle genes (Lascaris et al, 2003), was concomitantly downregulated more than eight-fold.
Figure 7.

Addition of glucose to a steady-state culture triggers an acceleration of the mRNA turnover. Effect of glucose pulse on expression of genes of the glycolytic, storage carbohydrate and TCA cycle metabolic pathways. The numbers represent the fold change calculated between the expression values obtained at 330 s and at the initial steady state (0 s). Green labels represent a downregulation and red labels represent an upregulation.
Our results are consistent with the notion that trehalose-6-phosphate (T6P) inhibition of glucose phosphorylation is required to avoid excessive phosphorylation and ‘glucose-accelerated death' (Blazquez et al, 1993; Francois and Parrou, 2001). The concentration of T6P increased by 15-fold within the first 180 s following the addition of glucose to reach a concentration (4.8 mM) (Figure 6I) that suffices for the complete in vitro inhibition of both hexokinase I (Ki=40 μM) and hexokinase II (Ki=200 μM) (Figure 6) (Blazquez et al, 1993). In the meantime, the genes GLK1, HXK1 and HXK2 encoding gluco- and hexokinases were also downregulated, thus reinforcing the notion that the cell limits glucose phosphorylation in response to a sudden increase in glucose availability.
The response of the metabolites of the upper part of the glycolysis was extremely rapid (within the first 30 s) and preceded all detectable transcriptional control. However, we also measured a significant increase in fructose-2,6-biphosphate (F2,6P2) about 120 s after the perturbation (Figure 6D). This rise was accompanied by a concomitant transcriptional upregulation of PFK27 (encoding a 6-phosphofructo-2-kinase (+2.74)) and a downregulation of PFK26 (encoding the second form of the 6-phosphofructose-2-kinase (−3.43)) and FBP26 (encoding the fructose-2,6-bisphosphatase (−1.8)), which is involved in the degradation of F2,6P2 (Figure 7). The accumulation of F2,6P2 fitted with its role in activating the phosphofructokinase activity rate, whereas the substrates levels (F6P and ATP) were low, maintaining a high product (F1,6P2) concentration. This correlation between metabolite and related transcripts levels illustrates how complex and synergistic metabolic and transcriptional control are in fine-tuning metabolic pathway regulations to master the large changes in metabolite concentration.
Fast decay of downregulated transcripts indicates active mRNA degradation
The average half-life of yeast poly(A)+ mRNA in S. cerevisiae has previously been estimated around 30 min using a temperature-sensitive RNA-pol II mutant (Wang et al, 2002). Figure 8A shows a comparison of mRNA half-lives observed by Wang et al (2002) with those calculated from the present study (see Supplementary information 8a and 8b for calculation). In our experiments, simultaneous transcription and degradation may occur, which should lead to an underestimation of the presented mRNA decay constant. Nevertheless, the set of 565 downregulated transcripts displayed an order of magnitude faster decay with an average half-life of 4 min (Figure 8A). This suggests that active mRNA degradation, which has previously been described for SDH1, SDH2 and SUC2, affects large sets of genes involved in processes such as the TCA cycle and storage carbohydrate metabolism. For example, 18 genes involved in the latter process (TPS1, TPS2, TSL1, ATH1, NTH2, GSY1, GSY2, GLG1, GLC3, GAC1, GPH1, GDB1, PGM2, UGP1, GIP2 FSP2, PIG2, PIG1) showed a much faster decay than expected based on previous data on mRNA half-lives (Wang et al, 2002) (Supplementary information 8).
Figure 8.

(A) Scatter plot comparing mRNA half-life measured by Wang et al (2002) and the mRNA half-life calculated from the data of this study. The dashed line represents the average value of half-life. (B) Motifs identified in the 3′ untranslated regions of the 163 genes downregulated belonging to overrepresented functional categories ‘energy' and ‘metabolism'. (C) Significance of the representation of motif identified in the tested set.
In S. cerevisiae and higher eukaryotes, mRNA degradation can be initiated by poly(A) tail shortening (van Hoof and Parker, 2002). After poly(A) tail removal, mRNA degradation involves the decapping enzyme Dcp1p (LaGrandeur and Parker, 1998) and the 5′-to-3′ exonuclease Xrn1p (Heyer et al, 1995). This mechanism was indeed proposed for the faster decay of SHD1, SHD2 and SUC2 genes (Prieto et al, 2000). Additionally, 3′ degradation may occur, which involves the exosome, a complex of 3′-to-5′ exonucleases. In addition to the mRNA degradation, the exosome is involved in the processing of several RNA species. In yeast, the exosome is recruited via the fixation of Puf3p on AU-rich motif located in the 3′UTR of a gene (Olivas and Parker, 2000; Jackson et al, 2004).
Possible involvement of 3′ degradation was investigated by a systematic analysis of the 250 base pairs downstream of the stop codon of 163 downregulated genes belonging to the significantly overrepresented functional categories (Figure 2A). Four consensus motifs were found statistically overrepresented compared to their respective genome representation by binomial probability (Figures 8B and C). Three of them were close variations of the already described Puf3p motif (UGUANAUA). A fourth motif was found in a small subset of 19 genes. Out of the 163 genes tested, 116 genes harbored at least one of the four motifs, and 80 genes carried two or more elements (Supplementary information 9). The observed correlation between fast mRNA decay and the presence of conserved 3′UTR sequences supported a widespread involvement of active mRNA degradation in the fast response of S. cerevisiae to glucose. This mechanistic synergy results in an accelerated disappearance of translational substrate, which might be because of energy saving and optimizing the translational efficiency of newly transcribed mRNA. However, in the metabolic context described in this study, this mechanism could also be considered as a nucleotides salvage pathway. The RNA degradation recovery might be of importance regarding the fitness of a strain to adapt to rapid change in environment.
With the exception of the responses in purine and sulfur metabolism, many of the transcriptional events after the relief from glucose limitation have previously been linked to the TOR signal transduction pathway. In particular, the TOR pathway has been implicated in the regulation of mRNA turnover in S. cerevisiae (Albig and Decker, 2001) and in the expression of genes for ribosomal RNA and ribosomal proteins (Martin et al, 2004; Schawalder et al, 2004; Rudra et al, 2005). In mammalian cells, mTOR has been proposed to be a homeostatic ATP sensor (Dennis et al, 2001). Based on the transcript levels alone, this would have offered an attractive explanation for the observed upregulation of TOR targets after relief from glucose limitation. However, the metabolite data revealed that, in fact, intracellular ATP concentrations decreased after the glucose pulse. This observation underlines how simultaneous analysis at different information levels (transcriptome, metabolome) can improve interpretation of biological phenomena.
Conclusion
In the present study, we exploit the accurate control of chemostat cultures to generate reproducible perturbation experiments. Although this approach has been previously achieved to study the rapid dynamics of metabolite pools in S. cerevisiae (Theobald et al, 1997; Visser et al, 2004), this is the first time this approach has been integrated with simultaneous transcriptome analyses. Our data reveal a clear and sequential adaptation of vital cellular processes in response to a sudden relief of glucose limitation. The first significant changes in gene expression were only visible between 120 and 210 s and were restricted to specific functional categories (Figure 2A). The incorporation of transcription factor binding activity data provided a regulatory map that was in agreement with the overrepresented categories (Figure 2B). The nature of the metabolome and the transcriptome responses were highly correlated. Our results indicate that, upon relief of glucose limitation, yeast cells are confronted with several physiological stresses, including a significant decrease of the energy charge and AXP pool. At the same time, they are gearing up to accelerate growth as shown by the reprogramming of the transcription and translation machinery. Restoration of the cellular homeostasis was measurable at both metabolome and transcriptome levels. The early drop in cellular AXP pool was followed by the upregulation of genes involved in purine biosynthesis, C1 metabolism, sulfur assimilation and purine salvage (Figure 4). A significant increase of T6P was measured after the relief of glucose limitation, followed by the coordinated downregulation of the three hexokinase encoding genes (Figures 6 and 7), consistent with a response to prevent ‘glucose-accelerated death'. As the glucose concentration decreased, a major expression decrease of the genes involved in T6P synthesis was observed. Redox balancing appeared to involve a regulation of central carbon metabolism and, in particular, glycolysis. This regulation occurred both as an adjustment of metabolite and effector concentrations (e.g. F2,6P), adjustment of the expression of genes encoding TCA cycle enzymes and a tight control of mRNA turnover (synthesis and degradation) (Figures 7 and 8). Our results showed that the decay rate of downregulated transcripts was nine-fold faster than reported earlier, suggesting that mRNA degradation participates actively in the regulation of translation.
Dynamic stimulus response studies are a vital element in integrative systems biology. The present study illustrates how high-quality data can be generated by the use of tightly controlled cultivation conditions and appropriate analytical tools. Experiments that, in addition to transcriptome and metabolome data, include information at other relevant information levels (e.g. proteome, phosphoproteome and fluxome, references) will be essential to meet the longstanding challenge of cellular physiology/systems biology: to come to an integral understanding of the responses of living cells to their physical and chemical environment.
Materials and methods
Strains and growth conditions
Chemostat cultivation
S. cerevisiae (CEN PK 113-7D) was cultivated in an aerobic carbon-limited chemostat culture in a 7-l fermentor (Applikon, Schiedam, The Netherlands) with a working volume of 4 l on the adapted doubled mineral medium (Verduyn et al, 1992) with 27.1 g/l of glucose and 1.42 g/l of ethanol, to support a biomass concentration of about 15 g DW/l. The dilution rate was 0.05/h and the airflow rate was 200 l/h. Other fermentation parameters are a pH controlled at 5, a temperature controlled at 30°C, an overpressure of 0.3 bar, stirrer speed of 600 r.p.m. and dissolved oxygen higher than 70%.
Glucose pulse experiment
At the age of 140 h, the steady-state chemostat culture was perturbed by the addition of 20 ml of glucose solution (200 g/l) to the fermentor so that the residual glucose concentration was suddenly increased to about 1 g/l (5.56 mM). The glucose solution was rapidly injected by a pneumatic system (<1 s). Samples were taken prior to the glucose pulse (steady-state samples) and within 360 s after the perturbation.
Sampling methods
Metabolite sampling method
Sample for intracellular metabolite analysis was obtained by withdrawing 1 ml of broth from the fermentor by a rapid sampling set up (Lange et al, 2001) into 5 ml of 60% (v/v) methanol/water at −40°C to immediately quench the metabolic activities. The sample was then processed according to the intracellular sampling processing method described by Wu et al (2005) to give about 500 μl of intracellular metabolite solution that is ready for further analysis.
Sample for extracellular metabolite analysis was obtained following the method described by Mashego et al (2003).
Sampling for microarrays
Sampling of cells from chemostats, probe preparation and hybridization to Affymetrix Genechip® microarrays were performed as described previously (Piper et al, 2002) The results for the initial steady state, 30, 60, 120, 300 and 330 s were derived from at least two independently cultured replicates (for the number of replicates analyzed per time point, see Supplementary information 1). The time point at 210 s was derived from a single culture.
Microarray analysis
Data acquisition and analysis
Acquisition and quantification of array images and data filtering were performed using Affymetrix Gene Chip Operating System (GCOS). Before comparison, all arrays were globally scaled to a target value of 150 using the average signal from all gene features using GCOS. The complete set of .CEL files is deposited at Genome Expression Omnibus database (Barrett et al, 2005) (http://www.ncbi.nlm.nih.gov/geo) series accession number GSE3821. To eliminate insignificant variations, genes with values below 12 were set to 12 as described by Piper et al (2002). From the 9335 transcript features on the YG-S98 array, a filter was applied to extract 6383 yeast open reading frames, of which there were 6084 different genes. This discrepancy was owing to several genes being represented more than once when suboptimal probe sets were used in the array design. To represent the variation in replicate measurements, the coefficient of variation (mean deviation divided by the mean) (Supplementary information 1) was calculated as described previously (Boer et al, 2003).
For statistical analyses, Microsoft Excel running the significance analysis of microarrays (SAM Version 1.12) add-in was used (Tusher et al, 2001) for multiclass analysis. Genes were called significantly changed in expression using SAM with an expected median false discovery rate of 0.6%. Hierarchical clustering of the obtained sets of significantly changed expression levels was subsequently performed using Genespring Version 7.2 (Agilent Technologies Inc., Palo Alto, CA). Two main profiles (ascendent and descendent) were identified. K-means analysis of ascending and descending profiles gene subsets was performed using Genespring Version 7.2 (Agilent Technologies Inc., Palo Alto, CA).
For the statistical assessment of overrepresentation of MIPS functional categories (FUNCAT) (http://mips.gsf.de/projects/funcat) (Ruepp et al, 2004) and GO biological processes (http://www.geneontology.org/) (Eilbeck et al, 2005) in the SAM-identified transcripts, a test employing hypergeometric distribution, FunSpec (http://funspec.med.utoronto.ca/) (Robinson et al, 2002), was used using a P-value cutoff of 0.01 with a Bonferroni correction. The probability was calculated as follows: the P-value of observing z genes, belonging to the same functional category, is:
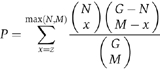
where N is the total number of genes in a functional category (Ruepp et al, 2004), M is the total number of genes in the cluster (upregulated clusters A, B, C and downregulated clusters D, E) and G is the total number of gene features on the YG98S array (6383).
The up- and downregulated data inspection for overrepresentation of transcription factors as defined by ChIP on chip analysis (http://jura.wi.mit.edu/fraenkel/download/release_v24/bound_by_factor/ORFs_bound_by_factor_v24.0.p005b_041213.txt) was also performed employing an in-house version of the hypergeometric distribution test. Applying the same formula, the probability was calculated where N is the total number of genes where the TF can bind upstream (Harbison et al, 2004), M is the total number of genes in the cluster (upregulated clusters A, B, C and downregulated clusters D, E) and G is the total number of gene features on the YG98S array (6383).
A search for conserved octa-nucleotide sequences in the 3′ untranslated region (250 nt) was performed using regulatory sequence analysis tools (van Helden et al, 2000) (http://rsat.scmbb.ulb.ac.be/rsat/). The occurrence of the discovered motif in the group of genes tested (163 genes) was compared with the expected occurrence of a group of same size randomly picked. The E-value represents the probability of finding the number of patterns with the same level of overrepresentation, which would be expected by chance alone. For instance, the E-value of a given motif is of the order of 10−6, indicating that, if we would submit random sequences to the program, such a level of overrepresentation would be expected every 1 000 000 trials. Motif structures were edited using the Weblogo program (Crooks et al, 2004). The sequence templates used to generate the Weblogo motifs are available as Supplementary information 9.
Analytical methods
Extracellular metabolites
The concentration of glucose and glycerol in the supernatant was measured with the Enzytec™ enzymatic kit (kit no. 1002781 for glucose, 1002809 for glycerol). The pyruvate concentration was measured by the Sigma Diagnostic kit (726-UV). The concentration of ethanol and acetic acid was measured by gas chromatography using a Chromopack CP 9001 with CP 9010 liquid sampler, connected to a Flame Ionisation Detector on an Innowax 15 m column (Hewlett Packard) with helium as the carrier gas.
Intracellular metabolites
Glycolytic intermediates (G6P, F6P, F1,6P2, F2,6P2, 2PG, 3PG, PEP), and TCA cycle intermediates (citrate, α-ketoglutarate, succinate, fumarate and malate), pentose phosphate pathway intermediate (6PG) and carbon storage intermediates (G1P, T6P) were analyzed by ESI-LC-MS/MS according to van Dam et al (2002) and the quantification was performed applying the isotope dilution LC-ESI-MS/MS (IDMS) method (Wu et al, 2005). In the case of F2,6P2, only peaks were measured instead of the absolute level and therefore the data are presented as the ratio to the steady-state condition.
NAD/NADH ratio was calculated by assuming that the lumped reaction catalyzed by aldolase, triphosphate-isomerase, glyceraldehyde-dehydrogenase, phosphoglycerate-kinase and phosphoglucomutase is close to equilibrium:
![]()
The NAD/NADH ratio is presented as the normalized value to the steady-state condition.
Nucleotide concentrations in the cell extract were analyzed by an ion pairing LC-ESI MS/MS method as described by Wu (2005) and quantified applying the IDMS method (Wu et al, 2005). All metabolite concentrations are provided in Supplementary information 5 and 7.
Calculation of mRNA half-life
mRNA degradation is modeled as
in which kd is the mRNA degradation constant, A is an additional model parameter to take into account measurement inaccuracy and tdelay is a time variable corresponding to the inflexion point of the transcript profile. A Matlab (The MathWorks Inc.) based nonlinear weighted least square program was developed to fit the above model parameters (A, kd) to the mRNA degradation profile, with the inverse variance of the measurements used as the weight. Furthermore, the mRNA half-life (t1/2) was calculated from the mRNA degradation constant (kd)
![]()
The data and program used for this calculation can be accessed in Supplementary information 8a and 8b, respectively. The results were compared with the mRNA half-life calculated by Wang et al (2002) available at the following URL: http://www-genome.stanford.edu/turnover/.
Supplementary Material
Supplemental Materials Content
Supplementary Data 1
Supplementary Data 2
Supplementary Data 3
Supplementary Data 4
Supplementary Data 5
Supplementary Data 6
Supplementary Data 7
Supplementary Data 8a
Supplementary Data 8b
Supplementary Data 9
Acknowledgments
The work performed in the Kluyver Centre for Genomics of Industrial Fermentation (Programs 1.07 and 5.5) was supported by the Netherlands Genomics Initiative.
References
- Albig AR, Decker CJ (2001) The target of rapamycin signaling pathway regulates mRNA turnover in the yeast Saccharomyces cerevisiae. Mol Biol Cell 12: 3428–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachellerie JP, Cavaille J (1997) Guiding ribose methylation of rRNA. Trends Biochem Sci 22: 257–261 [DOI] [PubMed] [Google Scholar]
- Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W, Edgar R (2005) NCBI GEO: mining millions of expression profiles—database and tools. Nucleic Acids Res 33: D562–D566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belde PJ, Vossen JH, Borst-Pauwels GW, Theuvenet AP (1993) Inositol 1,4,5-trisphosphate releases Ca2+ from vacuolar membrane vesicles of Saccharomyces cerevisiae. FEBS Lett 323: 113–118 [DOI] [PubMed] [Google Scholar]
- Blaiseau PL, Thomas D (1998) Multiple transcriptional activation complexes tether the yeast activator Met4 to DNA. EMBO J 17: 6327–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez MA, Lagunas R, Gancedo C, Gancedo JM (1993) Trehalose-6-phosphate, a new regulator of yeast glycolysis that inhibits hexokinases. FEBS Lett 329: 51–54 [DOI] [PubMed] [Google Scholar]
- Boer VM, de Winde JH, Pronk JT, Piper MD (2003) The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem 278: 3265–3274 [DOI] [PubMed] [Google Scholar]
- Buc PS, Rolfes RJ (1999) ade9 is an allele of SER1 and plays an indirect role in purine biosynthesis. Yeast 15: 1347–1355 [DOI] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA (2001) Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 12: 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghino GP, Atencio DP, Saghbini M, Beiner J, Scheffler IE (1995) Glucose-dependent turnover of the mRNAs encoding succinate dehydrogenase peptides in Saccharomyces cerevisiae: sequence elements in the 5′ untranslated region of the Ip mRNA play a dominant role. Mol Biol Cell 6: 1125–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghino GP, Scheffler IE (1996) Genetic analysis of glucose regulation in Saccharomyces cerevisiae: control of transcription versus mRNA turnover. EMBO J 15: 363–374 [PMC free article] [PubMed] [Google Scholar]
- Chapman AG, Atkinson DE (1977) Adenine nucleotide concentrations and turnover rates. Their correlation with biological activity in bacteria and yeast. Adv Microb Physiol 15: 253–306 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daran-Lapujade P, Jansen ML, Daran JM, van Gulik W, de Winde JH, Pronk JT (2004) Role of transcriptional regulation in controlling fluxes in central carbon metabolism of Saccharomyces cerevisiae. A chemostat culture study. J Biol Chem 279: 9125–9138 [DOI] [PubMed] [Google Scholar]
- Denis V, Boucherie H, Monribot C, Daignan-Fornier B (1998) Role of the myb-like protein bas1p in Saccharomyces cerevisiae: a proteome analysis. Mol Microbiol 30: 557–566 [DOI] [PubMed] [Google Scholar]
- Denis V, Daignan-Fornier B (1998) Synthesis of glutamine, glycine and 10-formyl tetrahydrofolate is coregulated with purine biosynthesis in Saccharomyces cerevisiae. Mol Gen Genet 259: 246–255 [DOI] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294: 1102–1105 [DOI] [PubMed] [Google Scholar]
- Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M (2005) The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol 6: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois J, Parrou JL (2001) Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 25: 125–145 [DOI] [PubMed] [Google Scholar]
- Gancedo JM (1998) Yeast carbon catabolite repression. Microbiol Mol Biol Rev 62: 334–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Werner-Washburne M (2002) The genomics of yeast responses to environmental stress and starvation. Funct Integr Genomics 2: 181–192 [DOI] [PubMed] [Google Scholar]
- Gelade R, Van de Velde S, Van Dijck P, Thevelein JM (2003) Multi-level response of the yeast genome to glucose. Genome Biol 4: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombert AK, Moreira dos SM, Christensen B, Nielsen J (2001) Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J Bacteriol 183: 1441–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert D, Philips PJ, Strange E (1971) Chemical analysis of microbial cells. In Methods in Microbiology, JR Norris, DW Ribbons (eds) Vol. 5B, pp 210–344. New York: Academic Press [Google Scholar]
- Heyer WD, Johnson AW, Reinhart U, Kolodner RD (1995) Regulation and intracellular localization of Saccharomyces cerevisiae strand exchange protein 1 (Sep1/Xrn1/Kem1), a multifunctional exonuclease. Mol Cell Biol 15: 2728–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskisson PA, Hobbs G (2005) Continuous culture—making a comeback? Microbiology 151: 3153–3159 [DOI] [PubMed] [Google Scholar]
- Jackson JS Jr, Houshmandi SS, Lopez LF, Olivas WM (2004) Recruitment of the Puf3 protein to its mRNA target for regulation of mRNA decay in yeast. RNA 10: 1625–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur TE, Parker R (1998) Isolation and characterization of Dcp1p, the yeast mRNA decapping enzyme. EMBO J 17: 1487–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange HC, Eman M, van Zullen G, Visser D, van Dam JC, Frank J, Teixeria de Mattos MH, Heijnen JJ (2001) Rapid sampling for intracellular metabolite determination. Biotech Bioeng 75: 406–415 [DOI] [PubMed] [Google Scholar]
- Lascaris R, Bussemaker HJ, Boorsma A, Piper M, van der SH, Grivell L, Blom J (2003) Hap4p overexpression in glucose-grown Saccharomyces cerevisiae induces cells to enter a novel metabolic state. Genome Biol 4: R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascaris RF, Groot E, Hoen PB, Mager WH, Planta RJ (2000) Different roles for abf1p and a T-rich promoter element in nucleosome organization of the yeast RPS28A gene. Nucleic Acids Res 28: 1390–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Cereghino GP, Scheffler IE (1992) Control of mRNA turnover as a mechanism of glucose repression in Saccharomyces cerevisiae. Mol Cell Biol 12: 2941–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN (2004) TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119: 969–979 [DOI] [PubMed] [Google Scholar]
- Mashego MR, van Gulik WM, Vinke JL, Heijnen JJ (2003) Critical evaluation of sampling techniques for residual glucose determination in carbon-limited chemostat culture of Saccharomyces cerevisiae. Biotechnol Bioeng 83: 395–399 [DOI] [PubMed] [Google Scholar]
- Mazon MJ, Gancedo JM, Gancedo C (1982) Phosphorylation and inactivation of yeast fructose-bisphosphatase in vivo by glucose and by proton ionophores. A possible role for cAMP. Eur J Biochem 127: 605–608 [DOI] [PubMed] [Google Scholar]
- Mercado JJ, Vincent O, Gancedo JM (1991) Regions in the promoter of the yeast FBP1 gene implicated in catabolite repression may bind the product of the regulatory gene MIG1. FEBS Lett 291: 97–100 [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Dirick L (1991) The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66: 995–1013 [DOI] [PubMed] [Google Scholar]
- Olivas W, Parker R (2000) The Puf3 protein is a transcript-specific regulator of mRNA degradation in yeast. EMBO J 19: 6602–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Heitman J (2000) Sok2 regulates yeast pseudohyphal differentiation via a transcription factor cascade that regulates cell–cell adhesion. Mol Cell Biol 20: 8364–8372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper MD, Daran-Lapujade P, Bro C, Regenberg B, Knudsen S, Nielsen J, Pronk JT (2002) Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J Biol Chem 277: 37001–37008 [DOI] [PubMed] [Google Scholar]
- Piper MD, Hong SP, Ball GE, Dawes IW (2000) Regulation of the balance of one-carbon metabolism in Saccharomyces cerevisiae. J Biol Chem 275: 30987–30995 [DOI] [PubMed] [Google Scholar]
- Prieto S, de la Cruz BJ, Scheffler IE (2000) Glucose-regulated turnover of mRNA and the influence of poly(A) tail length on half-life. J Biol Chem 275: 14155–14166 [DOI] [PubMed] [Google Scholar]
- Robinson MD, Grigull J, Mohammad N, Hughes TR (2002) FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics 3: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland F, Winderickx J, Thevelein JM (2002) Glucose-sensing and -signalling mechanisms in yeast. FEMS Yeast Res 2: 183–201 [DOI] [PubMed] [Google Scholar]
- Ronen M, Botstein D (2006) Transcriptional response of steady-state yeast cultures to transient perturbations in carbon source. Proc Natl Acad Sci USA 103: 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillon A, Barbey R, Patton EE, Tyers M, Thomas D (2000) Feedback-regulated degradation of the transcriptional activator Met4 is triggered by the SCF(Met30) complex. EMBO J 19: 282–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D, Zhao Y, Warner JR (2005) Central role of Ifh1p–Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J 24: 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Munsterkotter M, Mewes HW (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32: 5539–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schawalder SB, Kabani M, Howald I, Choudhury U, Werner M, Shore D (2004) Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432: 1058–1061 [DOI] [PubMed] [Google Scholar]
- Schuller HJ (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr Genet 43: 139–160 [DOI] [PubMed] [Google Scholar]
- Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, Daran JM, Pronk JT (2005) Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem 280: 437–447 [DOI] [PubMed] [Google Scholar]
- Theobald U, Mailinger W, Baltes M, Rizzi M, Reuss M (1997) In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae: I. Experimental observations. Biotechnol Bioeng 55: 305–316 [DOI] [PubMed] [Google Scholar]
- Theobald U, Mailinger W, Reuss M, Rizzi M (1993) In vivo analysis of glucose-induced fast changes in yeast adenine nucleotide pool applying a rapid sampling technique. Anal Biochem 214: 31–37 [DOI] [PubMed] [Google Scholar]
- Thevelein JM, Gelade R, Holsbeeks I, Lagatie O, Popova Y, Rolland F, Stolz F, Van de Velde S, Van Dijck P, Vandormael P, Van Nuland A, Van Roey K, Van Zeebroeck G, Yan B (2005) Nutrient sensing systems for rapid activation of the protein kinase A pathway in yeast. Biochem Soc Trans 33: 253–256 [DOI] [PubMed] [Google Scholar]
- Thomas D, Surdin-Kerjan Y (1997) Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 61: 503–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam JC, Eman MR, Frank J, Lange HC, van Dedem GWK, Heijnen SJ (2002) Analysis of glycolytic intermediates in Saccharomyces cerevisiae using anion exchange chromatography and electrospray ionization with tandem mass spectrometric detection. Anal Chim Acta 460: 209–218 [Google Scholar]
- van Dijken JP, Bauer J, Brambilla L, Duboc P, Francois JM, Gancedo C, Giuseppin MLF, Heijnen JJ, Hoare M, Lange HC, Madden EA, Niederberger P, Nielsen J, Parrou JL, Petit T, Porro D, Reuss M, van Riel N, Rizzi M, Steensma HY, Verrips CT, Vindelov J, Pronk JT (2000) An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Technol 26: 706–714 [DOI] [PubMed] [Google Scholar]
- van Helden J, Andre B, Collado-Vides J (2000) A web site for the computational analysis of yeast regulatory sequences. Yeast 16: 177–187 [DOI] [PubMed] [Google Scholar]
- van Hoof A, Parker R (2002) Messenger RNA degradation: beginning at the end. Curr Biol 12: R285–R287 [DOI] [PubMed] [Google Scholar]
- Verduyn C, Postma E, Scheffers WA, van Dijken JP (1992) Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8: 501–517 [DOI] [PubMed] [Google Scholar]
- Visser D, van Zuylen GA, van Dam JC, Eman MR, Proll A, Ras C, Wu L, van Gulik WM, Heijnen JJ (2004) Analysis of in vivo kinetics of glycolysis in aerobic Saccharomyces cerevisiae by application of glucose and ethanol pulses. Biotechnol Bioeng 88: 157–167 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO (2002) Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA 99: 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RM, Primig M, Washburn BK, Winzeler EA, Bellis M, Sarrauste DM, Davis RW, Esposito RE (2002) The Ume6 regulon coordinates metabolic and meiotic gene expression in yeast. Proc Natl Acad Sci USA 99: 13431–13436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L (2005) Development and application of experimental and modeling tools for in vivo kinetic analysis in S. cerevisiae. PhD dissertation TU Delft ISBN: 90-9020021-5
- Wu L, Mashego MR, van Dam JC, Proell AM, Vinke JL, Ras C, van Winden WA, van Gulik WM, Heijnen JJ (2005) Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal Biochem 336: 164–171 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials Content
Supplementary Data 1
Supplementary Data 2
Supplementary Data 3
Supplementary Data 4
Supplementary Data 5
Supplementary Data 6
Supplementary Data 7
Supplementary Data 8a
Supplementary Data 8b
Supplementary Data 9