

Abstract
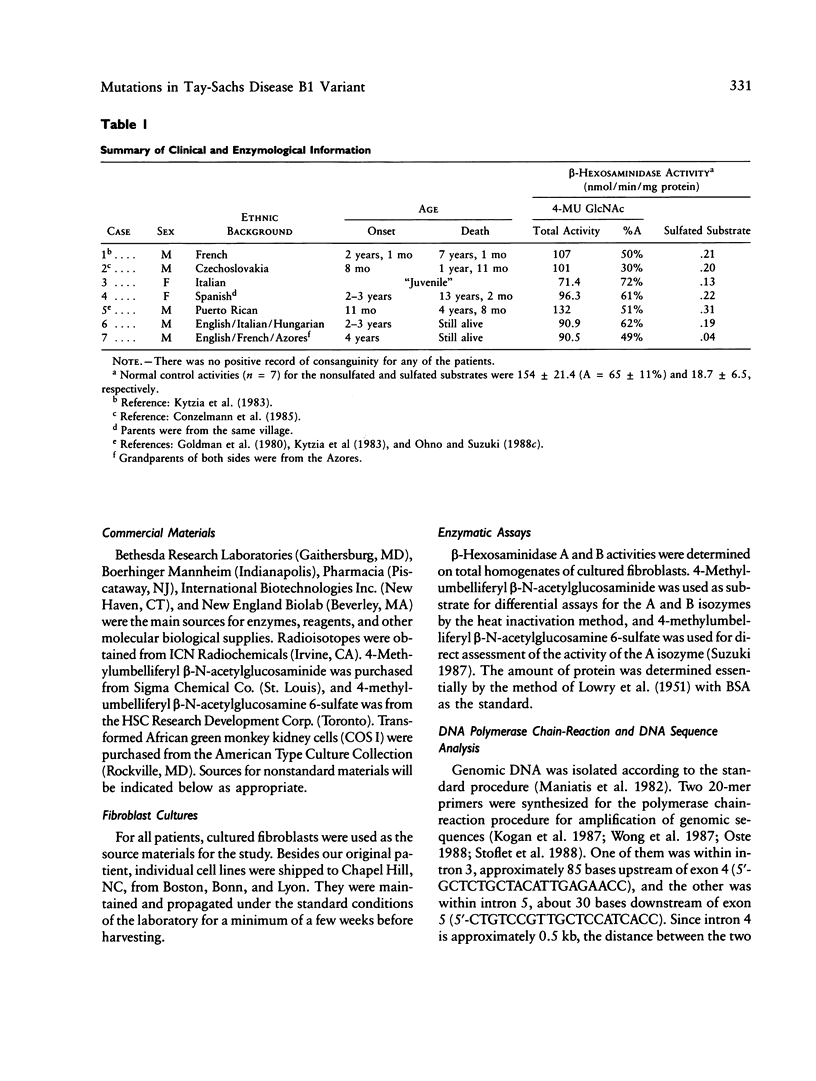
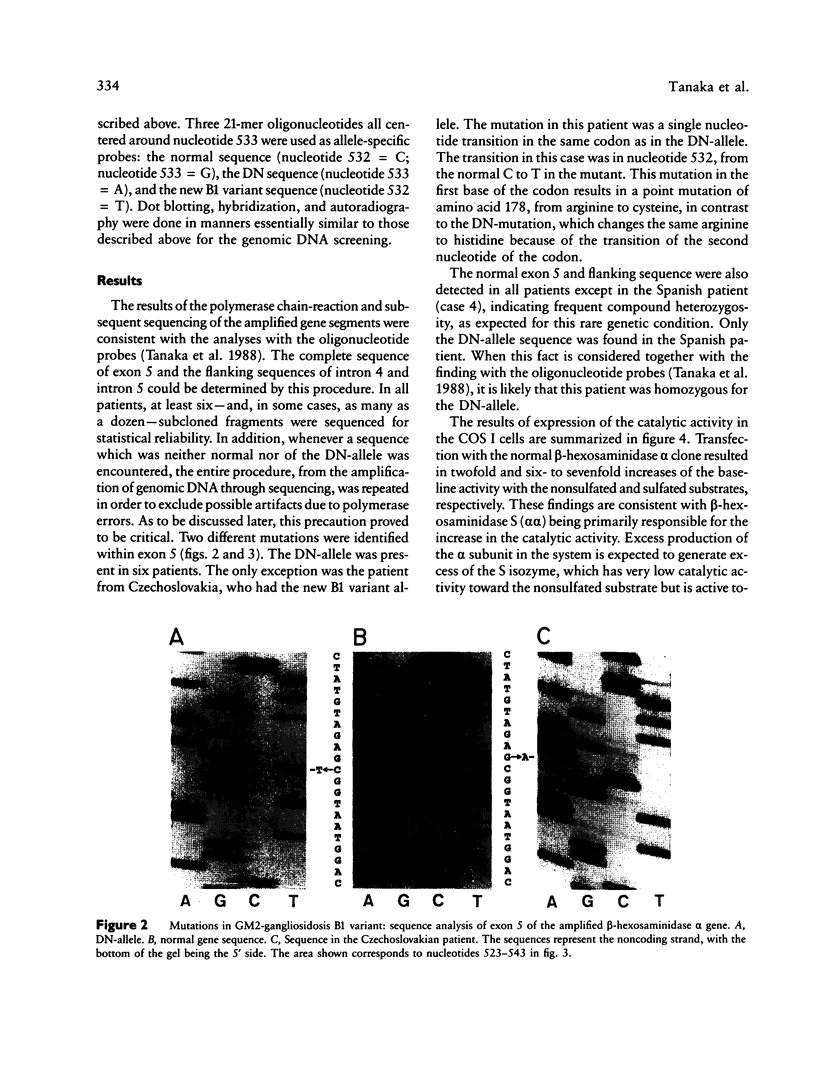
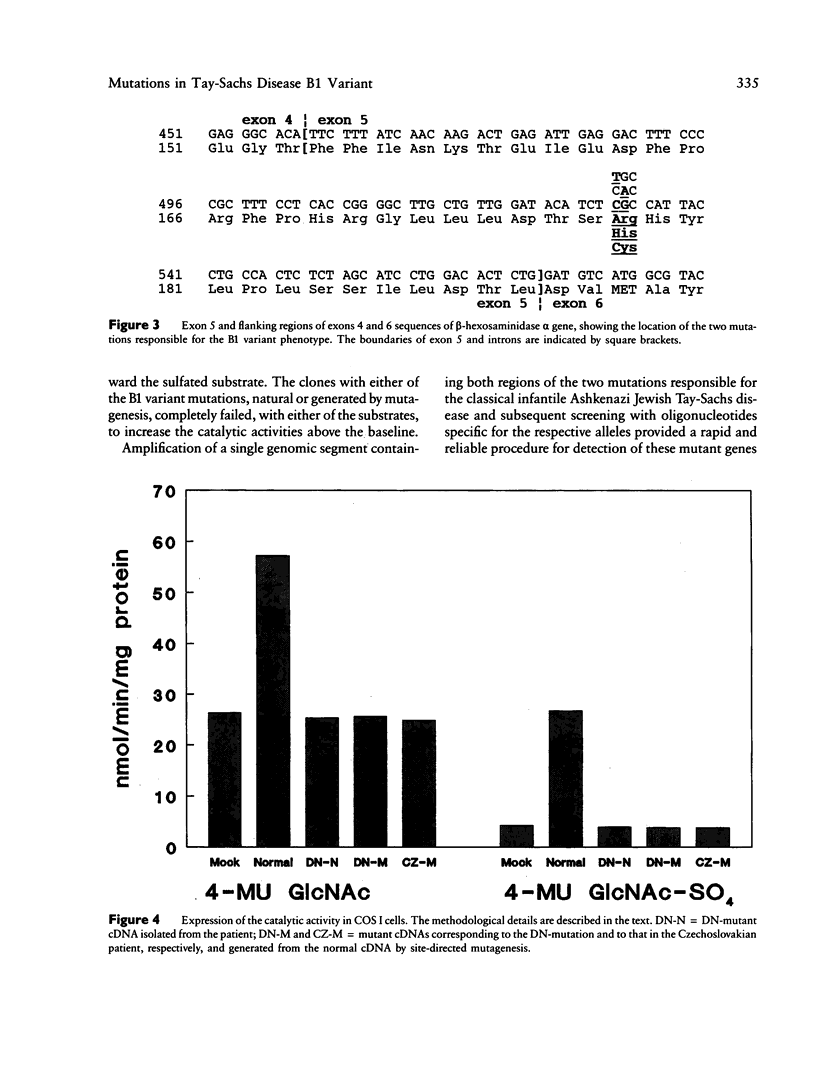
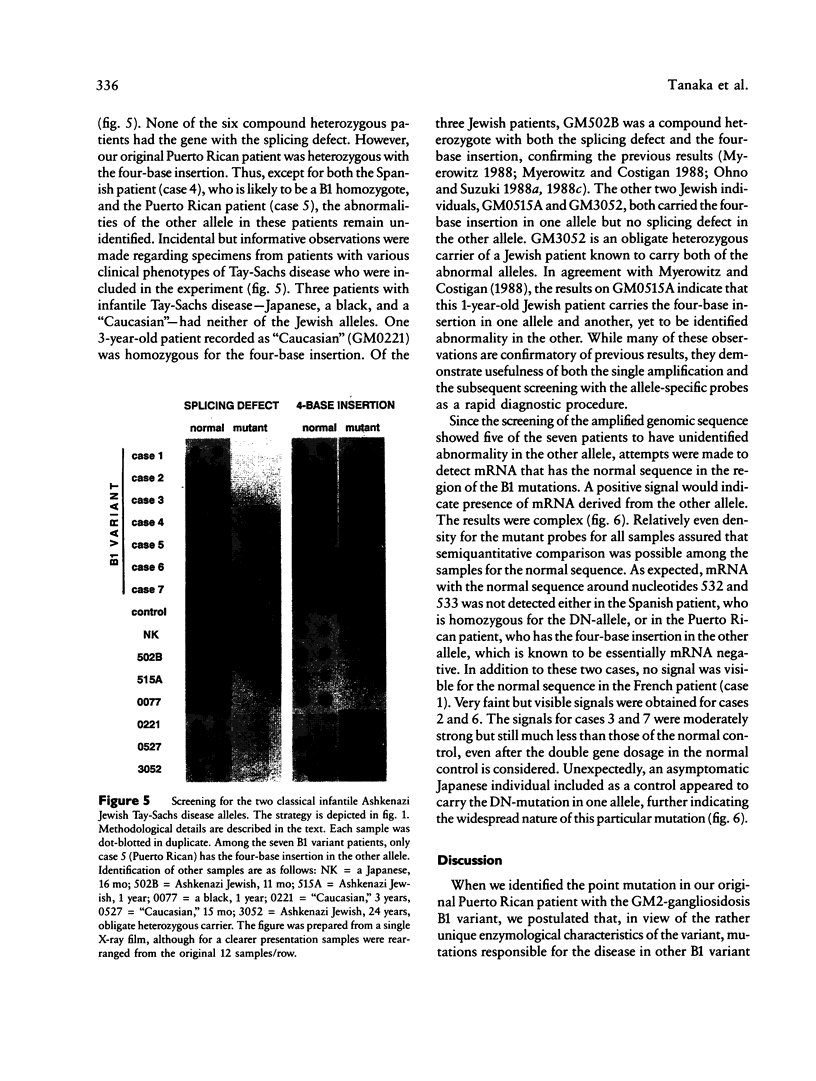
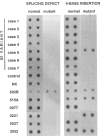
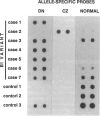
A single nucleotide transition within exon 5 of the beta-hexosaminidase alpha chain gene was identified in a Puerto Rican patient with GM2-gangliosidosis B1 variant as the mutation responsible for the unusual enzymological characteristics of this variant (G533----A; Arg178----His) (the DN-allele). A total of seven patients with enzymological characteristics of B1 variant have since been studied. They were Puerto Rican (DN), Italian, French, Spanish, two patients of mixed ethnic origin (English/Italian/Hungarian and English/French/Azores), and a Czechoslovakian. In confirmation of our earlier finding based on screening with allele-specific probes, all patients except the one from Czechoslovakia carried the same DN-allele. A new point mutation found in this patient changed the same codon affected in the DN-allele (C532----T; Arg178----Cys). An asymptomatic Japanese individual included as a control also carried one allele with the DN-mutation. Site-directed mutagenesis and expression studies in COS I cells demonstrated that either of the two point mutations abolishes the catalytic activity of the alpha subunit. The Spanish patient was homozygous for the DN-allele, but others were all compound heterozygotes. The Puerto Rican patient was a compound heterozygote with the DN-mutation in one allele and with the four-base insertion in exon 11, one of the two mutations found in the classical Ashkenazi Jewish Tay-Sachs disease, in the other allele. Abnormalities of the other allele were not identified in all other compound heterozygous patients. In these patients, the level of mRNA derived from the other allele was variable, ranging from being undetectable to being much lower than normal. This series of studies uncovered a new B1 variant mutation, confirmed our preliminary finding that the DN-allele has a surprisingly wide geographic and ethnic distribution, and pointed out the highly complex nature of the molecular genetics of this rare disorder. They also support our working hypothesis that mutations responsible for the unique enzymological characteristics of the B1 variant should be located in or near exon 5 of the gene and that this region of the enzyme protein is critical for its catalytic function.
Full text
PDF






Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Arpaia E., Dumbrille-Ross A., Maler T., Neote K., Tropak M., Troxel C., Stirling J. L., Pitts J. S., Bapat B., Lamhonwah A. M. Identification of an altered splice site in Ashkenazi Tay-Sachs disease. Nature. 1988 May 5;333(6168):85–86. doi: 10.1038/333085a0. [DOI] [PubMed] [Google Scholar]
- Biggin M. D., Gibson T. J., Hong G. F. Buffer gradient gels and 35S label as an aid to rapid DNA sequence determination. Proc Natl Acad Sci U S A. 1983 Jul;80(13):3963–3965. doi: 10.1073/pnas.80.13.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann E., Nehrkorn H., Kytzia H. J., Sandhoff K., Macek M., Lehovský M., Elleder M., Jirásek A., Kobilková J. Prenatal diagnosis of GM2 gangliosidosis with high residual hexosaminidase A activity (variant B1; pseudo AB variant). Pediatr Res. 1985 Nov;19(11):1220–1224. doi: 10.1203/00006450-198511000-00022. [DOI] [PubMed] [Google Scholar]
- Cullen B. R. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- Goldman J. E., Yamanaka T., Rapin I., Adachi M., Suzuki K., Suzuki K. The AB-variant of GM2-gangliosidosis. Clinical, biochemical, and pathological studies of two patients. Acta Neuropathol. 1980;52(3):189–202. doi: 10.1007/BF00705807. [DOI] [PubMed] [Google Scholar]
- Kogan S. C., Doherty M., Gitschier J. An improved method for prenatal diagnosis of genetic diseases by analysis of amplified DNA sequences. Application to hemophilia A. N Engl J Med. 1987 Oct 15;317(16):985–990. doi: 10.1056/NEJM198710153171603. [DOI] [PubMed] [Google Scholar]
- Kytzia H. J., Hinrichs U., Maire I., Suzuki K., Sandhoff K. Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity. EMBO J. 1983;2(7):1201–1205. doi: 10.1002/j.1460-2075.1983.tb01567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytzia H. J., Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985 Jun 25;260(12):7568–7572. [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Li S. C., Hirabayashi Y., Li Y. T. A new variant of type-AB GM2-gangliosidosis. Biochem Biophys Res Commun. 1981 Jul 30;101(2):479–485. doi: 10.1016/0006-291x(81)91285-7. [DOI] [PubMed] [Google Scholar]
- Myerowitz R., Costigan F. C. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J Biol Chem. 1988 Dec 15;263(35):18587–18589. [PubMed] [Google Scholar]
- Myerowitz R., Hogikyan N. D. Different mutations in Ashkenazi Jewish and non-Jewish French Canadians with Tay-Sachs disease. Science. 1986 Jun 27;232(4758):1646–1648. doi: 10.1126/science.3754980. [DOI] [PubMed] [Google Scholar]
- Myerowitz R., Piekarz R., Neufeld E. F., Shows T. B., Suzuki K. Human beta-hexosaminidase alpha chain: coding sequence and homology with the beta chain. Proc Natl Acad Sci U S A. 1985 Dec;82(23):7830–7834. doi: 10.1073/pnas.82.23.7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T., Muscillo M., Ohno K., Hoffman A. J., Suzuki K. A point mutation in the coding sequence of the beta-hexosaminidase alpha gene results in defective processing of the enzyme protein in an unusual GM2-gangliosidosis variant. J Neurochem. 1988 Sep;51(3):984–987. doi: 10.1111/j.1471-4159.1988.tb01836.x. [DOI] [PubMed] [Google Scholar]
- Navon R., Proia R. L. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989 Mar 17;243(4897):1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- Ohno K., Suzuki K. Multiple abnormal beta-hexosaminidase alpha chain mRNAs in a compound-heterozygous Ashkenazi Jewish patient with Tay-Sachs disease. J Biol Chem. 1988 Dec 5;263(34):18563–18567. [PubMed] [Google Scholar]
- Oste C. Polymerase chain reaction. Biotechniques. 1988 Feb;6(2):162–167. [PubMed] [Google Scholar]
- Paw B. H., Kaback M. M., Neufeld E. F. Molecular basis of adult-onset and chronic GM2 gangliosidoses in patients of Ashkenazi Jewish origin: substitution of serine for glycine at position 269 of the alpha-subunit of beta-hexosaminidase. Proc Natl Acad Sci U S A. 1989 Apr;86(7):2413–2417. doi: 10.1073/pnas.86.7.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia R. L., Neufeld E. F. Synthesis of beta-hexosaminidase in cell-free translation and in intact fibroblasts: an insoluble precursor alpha chain in a rare form of Tay-Sachs disease. Proc Natl Acad Sci U S A. 1982 Oct;79(20):6360–6364. doi: 10.1073/pnas.79.20.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar G., Sommer S. S. RNA amplification with transcript sequencing (RAWTS). Nucleic Acids Res. 1988 Jun 10;16(11):5197–5197. doi: 10.1093/nar/16.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderfeld S., Brendler S., Sandhoff K., Galjaard H., Hoogeveen A. T. Genetic complementation in somatic cell hybrids of four variants of infantile GM2 gangliosidosis. Hum Genet. 1985;71(3):196–200. doi: 10.1007/BF00284572. [DOI] [PubMed] [Google Scholar]
- Stoflet E. S., Koeberl D. D., Sarkar G., Sommer S. S. Genomic amplification with transcript sequencing. Science. 1988 Jan 29;239(4839):491–494. doi: 10.1126/science.3340835. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Enzymatic diagnosis of sphingolipidoses. Methods Enzymol. 1987;138:727–762. doi: 10.1016/0076-6879(87)38063-2. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Rapin I., Suzuki Y., Ishii N. Juvenile GM2-gangliosidosis. Clinical variant of Tay-Sachs disease or a new disease. Neurology. 1970 Feb;20(2):190–204. doi: 10.1212/wnl.20.2.190. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Suzuki K. Partial deficiency of hexosaminidase component a in juvenile gm2-gangliosidosis. Neurology. 1970 Sep;20(9):848–851. doi: 10.1212/wnl.20.9.848. [DOI] [PubMed] [Google Scholar]
- Tanaka A., Ohno K., Suzuki K. GM2-gangliosidosis B1 variant: a wide geographic and ethnic distribution of the specific beta-hexosaminidase alpha chain mutation originally identified in a Puerto Rican patient. Biochem Biophys Res Commun. 1988 Oct 31;156(2):1015–1019. doi: 10.1016/s0006-291x(88)80945-8. [DOI] [PubMed] [Google Scholar]
- Wong C., Dowling C. E., Saiki R. K., Higuchi R. G., Erlich H. A., Kazazian H. H., Jr Characterization of beta-thalassaemia mutations using direct genomic sequencing of amplified single copy DNA. 1987 Nov 26-Dec 2Nature. 330(6146):384–386. doi: 10.1038/330384a0. [DOI] [PubMed] [Google Scholar]