

Abstract
The activity of glutamate transporters is essential for the temporal and spatial regulation of the neurotransmitter concentration in the synaptic cleft, and thus, is crucial for proper excitatory signaling. Initial steps in the process of glutamate transport take place within a time scale of microseconds to milliseconds. Here we compare the steady-state and pre-steady-state kinetics of the neuronal heterologously expressed glutamate transporter EAAC1, cloned from the mammalian retina. Rapid transporter dynamics, as measured by using whole-cell current recordings, were resolved by applying the laser-pulse photolysis technique of caged glutamate with a time resolution of 100 μs. EAAC1-mediated pre-steady-state currents are composed of two components: A transport current generated by substrate-coupled charge translocation across the membrane and an anion current that is not stoichiometrically coupled to glutamate transport. The two currents were temporally resolved and studied independently. Our results indicate a rapid glutamate-binding step occurring on a submillisecond time scale that precedes subsequent slower electrogenic glutamate translocation across the membrane within a few milliseconds. The voltage-dependent steady-state turnover time constant of the transporter is about 1/10 as fast, indicating that glutamate translocation is not rate limiting. A third process, the transition to an anion-conducting state, is delayed with respect to the onset of glutamate transport. These rapid transporter reaction steps are summarized in a sequential shuttle model that quantitatively accounts for the results obtained here and are discussed regarding their functional importance for glutamatergic neurotransmission in the central nervous system.
The activity of high-affinity glutamate transporters is essential for the normal function of the mammalian central nervous system, where glutamate is the major excitatory neurotransmitter. Excitatory neurotransmission is initiated by the presynaptic release of glutamate into the synaptic cleft, where it binds to and activates ionotropic and metabotropic glutamate receptors. The degree of the postsynaptic response may be regulated by the concentration of glutamate present within the cleft as a function of time. This concentration is influenced not only by the rate of presynaptic release but also by the rate of diffusion and most importantly by glutamate transport. Indeed, high-affinity glutamate transporters have been localized to synaptic regions, and, to date, five distinct mammalian glutamate transporter subtypes have been cloned (reviewed in ref. 1).
Numerous functional studies of natively and heterologously expressed glutamate transporter subtypes showed that the transport process is electrogenic and presumably coupled to cotransport of three Na+ ions and one proton, and counter-transport of one K+ ion, per transported glutamate (2–4). In addition to the coupled transport current, substrate-induced chloride conductances have been associated with the activation of transporters (4, 5). This anion flux, however, is not linked stoichiometrically to glutamate transport (4).
Most of these transporter characteristics have been determined under steady-state conditions with a maximum time resolution of 100 ms. However, initial steps in the process of glutamate uptake, such as glutamate binding and early ionic movements, take place within a time scale of milliseconds or submilliseconds (6).
This study was designed to compare and contrast the properties of steady-state and pre-steady-state kinetics of the neuronal glutamate transporter EAAC1 (excitatory amino acid carrier), cloned from the mammalian retina. We report here the investigation of rapid transporter dynamics on a submillisecond time scale by applying the technique of laser-pulse photolysis of caged glutamate (7–9). The results are consistent with a sequential shuttle model, in which the electrogenic Na+-dependent glutamate translocation occurs within milliseconds and is preceded by submillisecond binding of glutamate to the transporter. Therefore, neuronal EAAC1 is capable of efficient and rapid removal of glutamate from the synaptic cleft during synaptic transmission.
Materials and Methods
Molecular Biology.
The coding sequence of the retinal EAAC1 glutamate transporter was isolated from complementary DNA (cDNA) derived from adult rat retina of both sexes by the polymerase chain reaction (PCR). Briefly, total RNA was isolated from rat retinae by the method of Chomczynski and Sacchi (10). Oligo(dT)-primed first-strand cDNA synthesis was performed with Superscript II reverse transcriptase (GIBCO/BRL). cDNA encoding the EAAC1 gene product was amplified from retinal first-strand cDNA by PCR using standard protocols (11). The sense and antisense primer sequences were 5′-CCATCATGGGGAAGCCCACG-3′ and 5′-AGTCCCAGGCATCTAAGGCC-3′, respectively. They correspond to the rat brain EAAC1 nucleotide sequences at positions 142–161 and 1724–1743 (12).
The PCR-generated EAAC1 insert included the 1569-bp coding sequence as well as 5 bp of 5′ and 29 bp of 3′ noncoding sequence. The amplification product was purified on an agarose gel, ligated into a EcoRV-digested pBluescript KS (+) plasmid vector (Stratagene), transformed into Escherichia coli XL1-Blue (Stratagene), sequenced on both strands (13), and subcloned into the modified expression vector pBK-CMV (Δ[1098–1300]) (Stratagene) at the BamHI and HindIII restriction sites. This construct was termed pCMV-EAAC1. Cell culturing (HEK293, ATCC no. CRL 1573) and transfections were performed as described (9, 14).
Whole-Cell Current Recording.
Glutamate-induced currents were measured in the whole-cell current-recording configuration at room temperature. The resistance of the recording electrode was 2-3 MΩ; the series resistance was 4-6 MΩ. Unless stated otherwise, the intracellular and extracellular solutions contained 130 mM KSCN, 1 mM MgCl2, 10 mM tetraethylammonium chloride, 10 mM EGTA, 10 mM Hepes (pH 7.3) and 140 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM glucose, 10 mM Hepes (pH 7.3), respectively. SCN− was in the intracellular solution because it enhances glutamate transporter-induced currents (4). Whole-cell currents were recorded with an Adams and List (AlaScientific Instruments, Westbury, NY) EPC7 amplifier under voltage-clamp conditions.
Laser-Pulse Photolysis and Rapid Solution Exchange.
Laser-pulse photolysis experiments were performed as described previously (9). Briefly, glutamate or α-carboxy-o-nitrobenzyl (αCNB)-caged glutamate (Molecular Probes) was applied with a U-tube device to the cells with a velocity of 5 cm/s and a time resolution of 20–30 ms (10–90% rise time with whole cells). Photolysis of caged glutamate was initiated with a light flash (340 nm, 15 ns, excimer laser pumped dye laser, Lambda Physik, Göttingen, Germany) which was delivered to the cell with an optical fiber (350-μm diameter). Laser energies were varied in the range of 50–400 mJ/cm2 with neutral density filters. To estimate the concentration of photolytically released glutamate, a standard glutamate concentration of 100 μM was applied to the cell by rapid perfusion before and after photolysis experiments, and the steady-state current amplitude was used to calculate the free glutamate concentration from the dose–response curve (9, 14).
Data were low-pass-filtered at 1–20 kHz, digitized with a sampling rate of 5–50 kHz, and recorded by using the pClamp6 software (Axon Instruments, Foster City, CA). The data were analyzed using the Origin (MicroCal, Northampton, MA) software. Kinetic modeling was performed with the Scientist program (Micromath, Salt Lake City, UT).
Results
Molecular Cloning and Functional Expression of EAAC1 in Mammalian Cells.
The neuronal glutamate transporter EAAC1 characterized here was isolated as cDNA (1,569 bp) from the mammalian retina. The predicted amino acid sequence comprises 523 amino acids and has a theoretical molecular mass of 56.7 kDa. It has 89% amino acid sequence identity with the rabbit EAAC1 (15), 90% identity with the human EAAT3 (16), and is identical to the rat brain EAAC1 (12).
Functional characterization of the retinal EAAC1 transporter subtype was performed by transient expression in HEK293 cells. Prior to transfection, the cells were analyzed for endogenous expression of the five known glutamate transporters subtypes (data not shown). Neither biochemical techniques—such as PCR, immunocytochemistry, immunoblot analysis (Fig. 1B, lane 1), and radiotracer experiments—nor electrophysiological experiments indicated the expression of endogenous EAAC1 in HEK293 cells.
Figure 1.
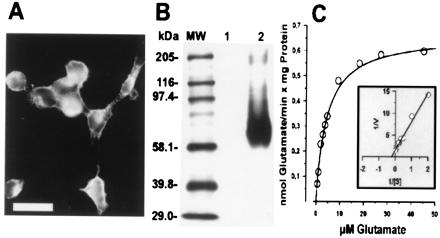
Protein expression and functional characterization of heterologously expressed EAAC1 in HEK293 cells. (A) Fluorescence micrograph of HEKEAAC1 cells immunolabeled for EAAC1. (Bar = 20 μm.) (B) Immunoblot analysis. Aliquots of SDS extracts (5 μg per lane) of membrane protein fractions of cells transfected with vector alone (lane 1) and HEKEAAC1 cells (lane 2) were subjected to SDS/10% polyacrylamide gel electrophoresis and were immunoblotted with EAAC1-specific antibodies (0.04 μg/ml). Molecular mass markers (lane MW) are indicated in kDa. (C) Kinetic properties of l-[3H]glutamate uptake into HEKEAAC1 membrane vesicle preparations. Uptake velocity was measured by using 1 μCi (1 μCi = 37 kBq) of l-[3H]glutamate and increasing concentrations of unlabeled l-glutamate at a membrane vesicle protein concentration of 40 μg. Measurements were taken within the linear range of uptake (15 and 30 s). The data are the mean of triplicate determinations from three experiments. The Km and Vmax values were obtained from the Lineweaver–Burk plot (Inset, n = 3).
Immunocytochemistry of pCMV-EAAC1-transfected HEK293 cells (HEKEAAC1) with EAAC1-specific antibodies revealed a membranous localization of the heterologously expressed transporter (Fig. 1A), whereas nontransfected or vector alone transfected HEK293 cells showed no staining either by immunocytochemistry or by immunoblot analysis (Fig. 1B, lane 1). As shown in Fig. 1B lane 2, immunoblot analysis demonstrated heterologous expression of the EAAC1 transporter protein with an electrophoretic mobility (centered at 69 kDa) similar to that determined for the native EAAC1 from rat retina (17). Consistent with our results, N-linked glycosylation of the native EAAC1 protein has been shown to increase its molecular mass by about 12 kDa (18, 19).
HEKEAAC1 cells mediated Na+-dependent uptake of glutamate (50 to 100-fold over background), whereas replacement of external Na+ ions by Li+ ions almost completely inhibited glutamate uptake. The l-[3H]glutamate uptake as a function of the glutamate concentration demonstrated first-order kinetics with a Km of 4.5 ± 0.6 μM (Fig. 1C), which is in the same range as determined for the native retinal glutamate uptake [Km = 2–5 μM (11)]. The Vmax value was dependent on the transfection efficiency and varied as much as 10-fold between experiments. The Km, however, was not affected by varying expression levels.
Glutamate-Dependent EAAC1 Currents.
The experiments described below compare the basic properties of EAAC1-mediated currents evoked by rapid perfusion of glutamate or photolytic release from caged glutamate. After rapid perfusion with l-glutamate (100 μM), whole-cell recordings of voltage-clamped HEKEAAC1 cells (V = 0 mV) revealed time-dependent inward currents that developed with a rapid onset (30 ms) and reached a steady-state level within 70 ms (Fig. 2A). Accordingly, photolytic release of glutamate (500 μM caged glutamate, 45 μM released) evoked a current with a similar amplitude, but additionally a rapidly decaying transient component was resolved (Fig. 2B). This current is characterized by a very fast onset (time constant 1/τrise = 530 ± 30 s−1) that is followed by a decay to the steady-state level with a time constant of 1/τdecay = 130 ± 30 s−1 (Fig. 2B). To a lesser extent the transient current component is also observed in rapid perfusion experiments with whole cells, but this current is too fast to be temporally resolved (Fig. 2A). However, using excised membrane patches with rapid perfusion of supersaturating substrate concentrations, Wadiche and Kavanaugh (20) previously reported the existence of pre-steady-state transient currents. Caged glutamate (500 μM) alone had no effect on EAAC1. Under both experimental conditions the steady-state and transient components of glutamate-induced currents required Na+ in the external medium (Fig. 2 A and B). The transient component was abolished after lowering the extracellular Na+ concentration to 20 mM, whereas the steady-state current was still present (Fig. 2B). In the absence of internal K+ ions the glutamate-induced current was abolished (not shown). However, when 5 mM internal K+, a value below the estimated affinity of the transporters for K+ ions (21, 22), was used the transient component of the glutamate-induced current was still present (Fig. 2C), indicating that this component is Na+ dependent but may not be K+ dependent. The steady-state component of the current was reduced to less than 20% of the maximum current (Fig. 2C), suggesting that EAAC1 turnover is slowed.
Figure 2.
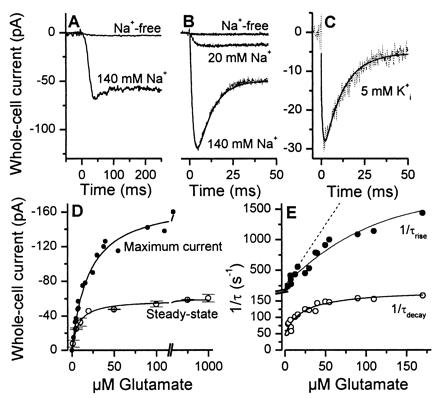
(A) Typical whole-cell current recording from an HEKEAAC1 cell. Glutamate (100 μM) was applied to the cell by rapid solution exchange in the absence (upper trace) and presence (lower trace) of 140 mM extracellular Na+ [V = 0 mV, KSCN-based intracellular solution, Na+ replaced by N-methyl-d-glucamine (NMG+)]. In this, and the following figures showing experiments in the absence of dl-threo-β-benzyloxyaspartate (TBOA) the leak current was subtracted. (B) Laser-pulse experiment with the same cell as in A at extracellular Na+ concentrations of 0, 20, and 140 mM. Glutamate was released from 500 μM α-carboxy-o-nitrobenzyl-caged glutamate with a 340-nm laser flash (180 mJ/cm2) at time 0. The solid line represents the best fit to the data according to the equation I(t) = −I∞ + Imaxexp(−t/τrise) − (Imax − I∞)exp(−t/τdecay). I represents current at times t (I(t)), t = ∞ (I∞), and at the maximum of the response (Imax). The time constants for the rise and the decay of the current were τrise = 1.9 ± 0.1 ms and τdecay = 8.0 ± 0.1 ms, respectively. The concentration of photolytically released glutamate was 45 ± 10 μM. (C) Similar laser-pulse experiment, but in the presence of only 5 mM intracellular K+ (K+ was replaced by NMG+, the intracellular anion was NO3−). The time constant τdecay was 9.8 ± 0.6 ms (fit shown as solid line). (D) The steady-state (○, rapid solution exchange) and maximum (●, laser-pulse photolysis) current is shown as a function of glutamate concentration (V = 0 mV, n = 4, error bars indicate ± SD). The line represents the best fit according to I = Imax(L/(L + Kapp))n (see text). (E) Glutamate concentration dependence of 1/τdecay (○) and 1/τrise (●) (V = 0 mV, four different cells). The lines represent the best fits according to Eqs. 2A and 2B (kop = 1400 s−1 and kcl = 630 s−1; see Results for the other parameters). The broken line represents a simulation of a simple bimolecular glutamate:transporter binding reaction with a slope of kd = 2⋅107 M−1⋅s−1 and k−d = 250 s−1.
The apparent Km value of the glutamate-induced steady-state currents (Kmss) in HEKEAAC1 was 5.1 ± 0.2 μM (Hill coefficient nH = 0.9 ± 0.1), in agreement with the radioactive uptake assay (Figs. 2D and 1C). The pre-steady-state currents, however, saturated at about 4-fold higher glutamate concentrations (Kmps = 20 ± 2 μM, Fig. 2D). In contrast to Kmss, which reflects the entire transport cycle (Eq. 1A), Kmps characterizes initial steps of the transporter reaction (Eq. 1B), suggesting that the intrinsic affinity for glutamate is lower than that estimated under steady-state conditions. This possibility is supported by the glutamate concentration dependence of 1/τdecay (Fig. 2E), which saturated with an apparent Km of 21 ± 8 μM, in accordance with Eq. 2A. Consistently, at subsaturating glutamate concentrations a glutamate-independent rate constant of 47 ± 18 s−1 was revealed (Eq. 2A). In addition, we determined the effect of the glutamate concentration on the rising phase of the current (Fig. 2E). The observed rate constant of the current rise, 1/τrise, is a nonlinear function of the glutamate concentration, indicating that it does not reflect only the bimolecular association process of glutamate with the transporter (Fig. 2E, broken line). Instead, this phase reflects the kinetics of the formation of the anion-conducting state (see also Fig. 3 A and B). In agreement with this interpretation, the concentration dependence of 1/τrise can be represented quantitatively by Eq. 2B (solid line, Fig. 2E). We calculated rate constants for the glutamate:EAAC1 binding/dissociation reaction of kd = 2⋅107 M−1⋅s−1 and kd = 1200 s−1, respectively, indicating that substrate binding and dissociation are in rapid pre-equilibrium with respect to glutamate translocation at concentrations higher than 10–15 μM, and that these processes are not rate limiting.
Figure 3.
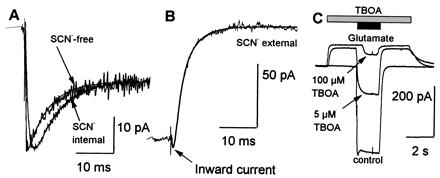
(A) Laser-pulse photolysis experiment in the absence of SCN− ([glutamate] = 60 ± 10 μM, V = 0 mV, SCN− was replaced by an equimolar concentration of CI−) (SCN−-free trace). The time constants were τrise = 0.2 ± 0.1 ms, and τdecay = 5.6 ± 0.4 ms. The SCN− internal trace shows, for comparison, a similar experiment in the presence of intracellular SCN− (τdecay = 6.3 ± 0.1 ms), which was rescaled (factor 0.22) to match the maximum amplitude of the two currents. (B) Laser-pulse photolysis experiment in the presence of extracellular SCN− (V = 0 mV, [glutamate] = 40 ± 10 μM, NaSCN-based extracellular solution, KCl-based intracellular solution). The time constant for the rise of the inward current were τ = 0.3 ± 0.1 ms, and for the rise of the outward current τ = 2.7 ± 0.6 ms (fit as in Fig. 2B). (C) Inhibition of the glutamate transporter by TBOA in the absence and presence of 25 μM glutamate. Bottom trace, control experiment in the absence of TBOA. The upper two traces show the same experiment after 2 s pre-incubation with 5 μM and 100 μM TBOA, respectively (V = 0 mV, KSCN-based intracellular solution, NaCl-based extracellular solution).
Dissection of Individual EAAC1-Associated Currents.
Glutamate transporter-associated currents are composed of glutamate-induced Na+/K+-coupled transport currents
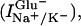 |
glutamate-induced uncoupled anion currents (IanionicGlu−), and glutamate-independent currents (Ianionic) (4, 20). Each component was isolated and separately analyzed.
Na+/K+-Coupled Currents
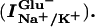 |
INa+/K+Glu− was isolated under pre-steady-state conditions by removing the electrochemical gradient for the anionic component as shown in Fig. 3A (SCN−-free trace). A concentration jump to 60 μM released glutamate evoked a current that exhibited a rapid transient phase. This transient phase relaxed to a steady-state level with a time constant of 1/τdecay = 180 ± 10 s−1 and a time constant for the rising phase of 1/τrise = 4500 ± 1000 s−1, suggesting a fast electrogenic reaction step early in the transport cycle which is not rate limiting. This fast time constant reflects a glutamate-induced reaction of EAAC1 and does not correspond to a process involved in the photolytic release of glutamate, because 1/τrise is 1/8 as fast as the release of glutamate from its caged precursor [1/τ(Photolysis) = 35,000 s−1 (8)].
Uncoupled Anion Currents (IanionicGlu−).
The contribution of IanionicGlu− to the total current was studied by varying the anionic electrochemical gradient. As shown in Fig. 3B, glutamate activation of EAAC1 in the presence of SCN− (4) in the extracellular solution ([SCN−]out ≫ [SCN−]in) caused an outward current which was preceded by a rapid inward current, characterizing the Na+/K+-transporter current component (Fig. 3B, arrowhead). Both components are kinetically distinct. Whereas the rise of INa+/K+Glu− occurred with a time constant of 1/τ = 3300 ± 800 s−1, the onset of IanionicGlu− was about 10-fold delayed (1/τ = 370 ± 100 s−1). A similar effect was revealed by inverting the anionic gradient ([SCN−]in ≫ [SCN−]out), resulting in an inward direction of both current components (INa+/K+Glu−) and IanionicGlu−); however, their onset was phase shifted (Fig. 3A). Thus, both experiments indicate that the activation of the EAAC1 anion-conducting state is delayed in comparison to the onset of glutamate translocation in the transport cycle.
Glutamate-Independent Currents.
The third component of the EAAC1-associated currents is glutamate independent and was dissected by the application of TBOA (Fig. 3C), a competitive and nontransported inhibitor (23). In the presence of glutamate (25 μM), TBOA inhibited the inward current (INa+/K+Glu− + IanionicGlu−) in a concentration-dependent manner with an apparent inhibition constant (Ki,app) of 4.9 ± 0.4 μM. In the absence of glutamate, however, TBOA generated a concentration-dependent outward current (Fig. 3C) with a Ki,app of 0.6 ± 0.1 μM, which is most likely caused by the inhibition of the glutamate-independent EAAC1 passive leakage current. This current is carried by anions, because it reverses close to 0 mV in the presence of symmetrical SCN− concentrations (Fig. 4D), in line with the behavior of other glutamate transporter subtypes (20).
Figure 4.
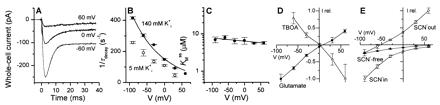
(A) Laser-pulse photolysis experiments at transmembrane potentials of −60, 0, and 60 mV ([glutamate] = 60 ± 10 μM, conditions and fitting as in Fig. 2B). The time constants are as follows: for −60 mV, τrise = 1.3 ± 0.1 ms, τdecay = 3.4 ± 0.1 ms; for 0 mV, τrise = 1.0 ± 0.1 ms, τdecay = 6.4 ± 0.1 ms; for 60 mV, τrise = 0.8 ± 0.3 ms, τdecay = 19 ± 7 ms. (B) Voltage dependence of 1/τdecay in the presence of 140 mM (●, [glutamate] = 60 μM) and 5 mM (○, [glutamate] = 50 μM) intracellular K+ (conditions of the experiments same as in A). Error bars indicate ± SD (n ≥ 3). (C) Voltage dependence of the apparent Kmss. The solid line was calculated according to Eq. 1 (Appendix) with Kd = 40 μM, kf0 = 210 s−1, kb0 = 40 s−1, Φ1 = 20, Φ2 = 0.5, and δ = 0.8. Error bars indicate ± SD (n ≥ 4). (D) Current–voltage relationship of the relative steady-state current [I(V)/|Imax∥] in the presence of 140 mM SCN− on both sides of the membrane evoked by 125 μM glutamate (●) or 5 μM TBOA (Δ). Error bars indicate ± SD (n ≥ 4) (E) Same experiment as in D, but in the absence of SCN− (▴) and in the presence of only extracellular SCN− (□), and only intracellular SCN− (○) (140 mM concentration, respectively). SCN− was replaced by equimolar amounts of chloride. Error bars indicate ± SD (n ≥ 4).
So far, the results obtained under pre-steady-state conditions suggest a fast electrogenic step early in the transport cycle which is not rate limiting. This step was further characterized by investigating the voltage dependence of the EAAC1 transport process.
Voltage Dependence of EAAC1 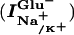 and IanionicGlu−.
and IanionicGlu−.
As shown in Figs. 2F and 4A and B, 1/τdecay was glutamate and voltage dependent (e-fold increase per −85 mV) and did not saturate within the voltage range examined. Although this decay was slightly slowed at subsaturating intracellular K+ concentrations (Eq. 2A), its voltage dependence was virtually unchanged (Fig. 4B, ○), indicating that the transient current component is not related to the K+-driven relocation of EAAC1. In contrast, this reaction is associated with a Na+-dependent transport step because the transient current was abolished in the absence of extracellular Na+ and 1/τdecay decreased with decreasing Na+ concentrations (Fig. 2B). Furthermore, the decay was accelerated by negative transmembrane potentials, but showed neither a maximum nor a minimum as a function of voltage, which indicates a nonreversible reaction step. Finally, this process was also observed under homoexchange conditions in the presence of symmetrical Na+ and glutamate concentrations and the absence of intracellular K+ (data not shown). All these observations together suggest that this process is closely associated with the quasi-irreversible Na+-driven glutamate translocation step, assuming zero-trans conditions. In contrast to a previous report (24), our results suggest that this initial step of glutamate translocation is not rate limiting. To test this idea, the voltage dependence of the stationary component of INa+/K+Glu− was determined. INa+/K+Glu− was inwardly directed, increased exponentially with decreasing membrane potentials (e-fold per −70 mV) and did not reverse within the voltage range examined (Fig. 4E, ▴). The latter effect is typical for zero-trans conditions as employed in this experiment. Additionally, INa+/K+Glu− did not saturate at negative membrane potentials in the presence of saturating glutamate concentrations, which is in agreement with previous reports (24). Consequently, our results suggest a second voltage-sensitive transporter reaction step which is ultimately the rate-limiting step in the EAAC1 transport cycle and determines the transporter properties at steady state. Kmss is affected by the kinetics of both electrogenic reaction steps (Eq. 1) and, thus, is expected to be voltage independent, assuming that the electrogenicity of both electrogenic reaction steps is equal and opposite (see below) and neglecting other electrogenic reaction steps, such as Na+ binding. In fact, as shown in Fig. 4C, Kmss is virtually voltage independent. In this context it is important to note that extracellular binding of one Na+ ion was proposed to be weakly voltage dependent (25). The experiments performed here do not permit testing this proposal, therefore, the voltage dependence of this step was not included in the model. Finally, the effect of SCN− on the voltage dependence of glutamate-induced currents was determined. In the presence of SCN− in the intracellular solution (Fig. 4E, □, [SCN−]out = 0), the current did not reverse as predicted for an infinite electrochemical driving force of SCN−. However, in the absence of a transmembrane concentration gradient for SCN− the current was always inwardly directed at 0 mV and reversed at +8 mV and not at the expected value of −2 mV (Fig. 4D, ●; [SCN−]in ≈ [SCN−]out), indicating that this current is partly carried by INa+/K+Glu−. The effect was strongly pronounced when SCN− was present in the external solution. Under these conditions, the reversal potential shifted to −52 mV (Fig. 4E, ○, [SCN−]in = 0), and the inwardly directed current at V < −52 mV predominately represents INa+/K+Glu−.
Discussion
Here, we show the application of the laser-pulse photolysis technique using caged glutamate to investigate rapid pre-steady-state kinetics of glutamate transporters. The time resolution of this method, in the range of 100 μs (9), is sufficient to temporally separate individual transporter reaction steps. Furthermore, this method will supplement the use of voltage-jump experiments (25) to study glutamate transporter pre-steady-state kinetics.
Under stationary conditions, the voltage dependence, Na+ and K+ dependence, as well as the pharmacology of glutamate-evoked currents in heterologously expressed EAAC1 from retina are similar to those from their brain counterparts (16, 23, 24). Glutamate-induced currents mediated by the retinal EAAC1 consist of two components, the electrogenic uptake current (INa+/K+Glu−) and an associated anion current (IanionicGlu−), consistent with previous studies (4).
The rapid kinetic analysis of the EAAC1 currents exposed four distinguishable phases that reflect distinct reaction steps of the transport cycle: the glutamate-binding step, the transition to the anion-conducting state, the glutamate-translocation step, and the formation of a glutamate-independent anion-conducting state. The simplest interpretation consistent with these observations is a sequential shuttle transport model, illustrated in Fig. 5A. In analogy to models previously proposed (5, 20, 22), our model incorporates a glutamate-binding step (characterized by the intrinsic glutamate dissociation constant Kd) that is followed by the voltage- and Na+-dependent but K+-independent glutamate translocation step across the membrane. Additionally, two anion-conducting states are included for the glutamate-induced anion current and the glutamate-independent leakage current.
Figure 5.
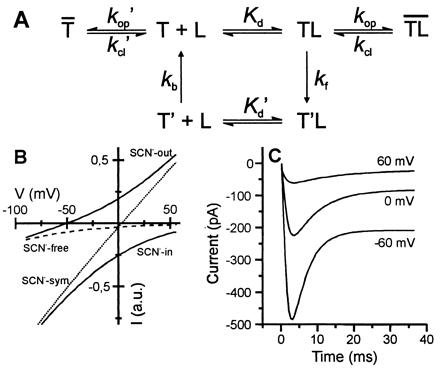
(A) Kinetic model for glutamate transport by EAAC1. Glutamate binding to the empty transporter, T, leads to the formation of the glutamate-bound state, TL. T′ and T′L are the respective states with the substrate binding site exposed to the cytoplasm. Charge translocation is assumed to be quasi-irreversible (zero-trans conditions). Na+ and K+ binding steps were assumed to be in rapid pre-equilibrium. (B) Simulation of steady-state anion and transport currents (Eqs. 3 and 4, Appendix). The following parameters were used: Kd = 50 μM, kf0 = 300 s−1, kb0 = 40 s−1, Φ1 = 20, Φ2 = 0.5, γ = 4 fS, z = −2, δ = 0.8. The conditions of the simulation were chosen similar to the experiments shown in Fig. 4 D and E. (C) Simulation of voltage-dependent currents as a function of time by numerical integration of the rate equations pertaining to the mechanism in A (conditions as in Fig. 4A). The rate constants for glutamate binding and dissociation were set to 2⋅107 M−1⋅s−1 and 1000 s−1, and kop and kcl were set to 1400 s−1 and 700 s−1, respectively. The other parameters were identical to those in B.
According to our results, equilibration of the glutamate-binding step occurs within less than 200 μs under physiological glutamate concentrations and precedes subsequent slower transporter reaction steps. Thus, the bimolecular glutamate-binding rate constant (≈2⋅107 M−1⋅s−1) of EAAC1 is consistent with that determined for ionotropic glutamate receptors (26). Furthermore, the intrinsic affinity of the transporter for glutamate was estimated to be less than 1/4 of that determined under steady-state conditions. The formation of the glutamate-gated anion-conducting state is delayed with respect to glutamate binding and occurs in a range of 0.5 to 5 ms. The glutamate-binding step is followed by a voltage- and Na+-dependent reaction step that takes place within 5–20 ms (rate constant kf, Fig. 5A). We propose that this reaction reflects the Na+-driven glutamate translocation step but not the extracellular Na+ binding or intracellular Na+ dissociation for two reasons: (i) A voltage-sensitive external Na+-binding reaction would result in a saturation of 1/τdecay at very negative potentials and (ii) rapid voltage-sensitive intracellular Na+ dissociation would result in a voltage independence of 1/τdecay. Both scenarios are inconsistent with our experimental data. However, at present we cannot rule out that a slow intracellular Na+ dissociation reaction is responsible for the voltage dependence of 1/τdecay. To keep the kinetic scheme as simple as possible, we have included all these reactions in one electrogenic quasi-irreversible reaction step which is characterized by the rate constant kf (Fig. 5A). This process is accelerated by negative transmembrane potentials and is not rate limiting as demonstrated by the rapid transient current observed either in the absence or in the presence of anions (SCN−) as well as in the presence of a low concentration of intracellular K+. If the glutamate translocation step would be rate limiting, as proposed by Kanai and colleagues (24), a voltage-dependent rise of the current to a steady-state level would be expected and not a transient current signal, which is demonstrated here. Thus, these results suggest the existence of a second voltage-dependent transporter reaction step late in the transport cycle that is rate limiting (rate constant kb, Fig. 5A).
The stoichiometry of glutamate transporters was proposed as cotransport of 1 glutamate/3 Na+/1 H+ and countertransport of 1 K+ ion (3). On the basis of this stoichiometry, the second rate-limiting electrogenic step may be associated either with the Na+ dissociation on the cytoplasmic side or with the relocation of the K+-bound transporter. Since rate constants of Na+-binding and dissociation for transporters, such as the Na+/Ca2+-exchanger (27) and Na+-complexing organic ligands (28), are typically rapid, we propose that for EAAC1 either the relocation of the K+-bound transporter or a conformational change induced by dissociation of Na+ is rate limiting. In the first case the countercharge of the unloaded transporter must be more negative than −1. Consistent with this, Kmss is virtually voltage independent.
In this study, a kinetic model is developed that quantitatively describes the binding of glutamate, the translocation of substrate, the translocation of coupled ions, and the formation of anion-conducting states. Predictions of time-dependent and voltage-dependent kinetics by numerical integration (Fig. 5) are in agreement with our experimental data. The model predicts an intrinsic Kd for glutamate of 50 μM. The forward rate constant for glutamate translocation at 0 mV is kf = 300 s−1. The turnover rate of the transporter is calculated to be 30 s−1 at 0 mV transmembrane potential and increases e-fold per −66 mV. The product of Po, which is the fraction of transporter in the anion-conducting state, and the single-channel conductance, γ, was estimated to be roughly 0.3 fS in the presence of SCN− at 0 mV. This conductance value indicates a rate constant for anion movement across the membrane that is consistent with either a carrier or an ion-channel model (29). To differentiate between these possibilities, a more detailed analysis is necessary. The low value of P0⋅γ estimated here, however, explains the inability to measure single-channel currents of EAAC1.
EAAC1 is localized postsynaptically in both brain and retina (17, 18). In the retina, EAAC1 is expressed in different neuronal cells (horizontal, cone bipolar, amacrine, and ganglion cells) and might be directly involved in the synaptic transmission process (11, 17). During glutamatergic transmission, neuronal glutamate transporters are in a depolarized environment which is expected to reduce the transport rate dramatically. We propose here for EAAC1 a unique voltage-dependence that counteracts this effect. The two positive charges that are translocated together with glutamate are distributed over several reaction steps of the transport cycle. Under depolarized conditions, this distribution may prevent excessive slowdown of initial steps in the transport cycle, such as the Na+-driven glutamate translocation step, whereas after repolarization a high stationary glutamate concentration gradient will be maintained.
Under physiological transmembrane potentials, the turnover rate of EAAC1 was estimated here as 90–110 s−1, whereas that estimated for the glial transporter EAAT2 is 15 s−1 (25). Even though it is severalfold faster, the turnover rate of EAAC1 is insufficient to account for rapid glutamate removal during synaptic transmission. Therefore, it has been suggested that transporters initially remove free glutamate from the synaptic cleft by binding rather than direct uptake (6, 25). However, this hypothesis requires that the residency time of glutamate binding at the transporter is longer than the synaptic response (≥1 ms) to avoid dissociation and further activation of receptors. The time constant of 0.2–0.3 ms for glutamate binding to EAAC1, however, suggests that glutamate binding is reversible during a typical synaptic event, indicating that the minimum glutamate concentration achievable by buffering would be in the range of 50 μM, even with a 2-fold excess of transporter binding sites over glutamate molecules in the synaptic cleft. Thus, another mechanism is required; it is provided by the quasi-irreversible rapid glutamate translocation step (≈3 ms) preceding the relatively slow stationary transporter turnover (≈10 ms). Therefore, it appears likely that the decay of the glutamate concentration at the synapase is at least biphasic, governed at first by reversible buffering, which is followed by rapid glutamate translocation and finally diffusion out of the synaptic cleft.
While this article was in the review process a study was published concerning the pre-steady-state kinetics of another glutamate transporter subtype (EAAT2); its conclusions are partly similar (30).
Acknowledgments
We thank M. Dumbsky for excellent technical assistance, E. Bamberg and H. Wässle for continuous encouragement and support, and E. Bamberg, A. Hirano, and K. Fendler for critically reading and improving the manuscript. We thank K. Shimamoto for kindly providing TBOA. This work was supported by the Deutsche Forschungsgemeinschaft (Grant 1393/2-1 awarded to C.G.).
Abbreviation
- TBOA
dl-threo-β-benzyloxyaspartate
Appendix
According to the kinetic model shown in Fig. 5A, Kmss and Kmps can be given as
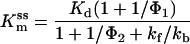 |
1A |
 |
1B |
Here, Φ1 and Φ2 are the equilibrium constants for formation of the anion-conducting states in the absence and presence of glutamate, respectively.
The observed rate constant of the decay of the transient current can be expressed as follows (L = [glutamate]):
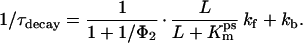 |
2A |
This equation was derived using the assumption that glutamate binding and formation of the anion-conducting states are in rapid pre-equilibrium. kf and kb represent the rate constants of glutamate translocation and transporter relocation, respectively. Assuming that the glutamate translocation can be neglected during the formation of the anion-conducting state (Fig. 2E), the rate constant for 1/τrise can be expressed as follows:
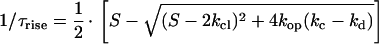 |
2B |
with S = L⋅kd + k−d + kop + kc. Here, kd and k−d are the rate constants for glutamate binding and dissociation and kop and kcl are the rate constants for formation and deactivation of the anion-conducting state, respectively.
The steady-state current in the absence and presence of SCN− can be expressed as (T0 is the number of transporters under observation, and e is the elementary charge; it was assumed that both the charge translocation and the relocation step are electrogenic):
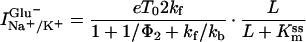 |
3 |
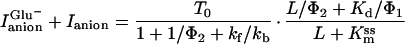 |
4 |
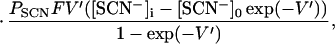 |
where V′ = FV/RT and PSCN = γRT⋅NA/(F2[SCN−]). F is the Faraday constant, T the temperature, R the gas constant, V the transmembrane potential, PSCN the single-channel permeability, γ the single-channel conductance, and NA the Avogadro constant. The voltage dependence of the current and 1/τdecay (Eq. 2A) is reflected by the voltage-dependent rate constants kf = kf0 exp(−zδV′/2) and kb = kb0 exp(zδV′/2), where kf0 and kb0 are the rate constants at V = 0 mV, z is the number of charges moving in the electric field, and δ is the fraction of the electric field sensed by the respective process.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160170397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160170397
References
- 1.Fairman W A, Amara S G. Am J Physiol. 1999;277:F481–F486. doi: 10.1152/ajprenal.1999.277.4.F481. [DOI] [PubMed] [Google Scholar]
- 2.Kanner B I, Sharon I. Biochemistry. 1978;17:3949–3953. doi: 10.1021/bi00612a011. [DOI] [PubMed] [Google Scholar]
- 3.Zerangue N, Kavanaugh M P. Nature (London) 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 4.Wadiche J I, Amara S G, Kavanaugh M P. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 5.Otis T S, Jahr C E. J Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diamond J S, Jahr C E. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hess G P. Biochemistry. 1993;32:989–1000. doi: 10.1021/bi00055a001. [DOI] [PubMed] [Google Scholar]
- 8.Wieboldt R, Gee K R, Niu L, Ramesh D, Carpenter B K, Hess G P. Proc Natl Acad Sci USA. 1994;91:8752–8756. doi: 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu L, Grewer C, Hess G P. In: Techniques in Protein Chemistry. Marshak D R, editor. Vol. 7. San Diego: Academic; 1996. pp. 139–149. [Google Scholar]
- 10.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Rauen T, Taylor W R, Kuhlbrodt K, Wiessner M. Cell Tissue Res. 1998;291:19–31. doi: 10.1007/s004410050976. [DOI] [PubMed] [Google Scholar]
- 12.Bjoras M, Gjesdal O, Erickson J D, Torp R, Levy L M, Ottersen O P, Degree M, Storm-Mathisen J, Seeberg E, Danbolt N C. Brain Res Mol Brain Res. 1996;36:163–168. doi: 10.1016/0169-328x(95)00279-2. [DOI] [PubMed] [Google Scholar]
- 13.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grewer C. Biophys J. 1999;77:727–738. doi: 10.1016/S0006-3495(99)76927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanai Y, Hediger M A. Nature (London) 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 16.Arriza J L, Fairman W A, Wadiche J I, Murdoch G H, Kavanaugh M P, Amara S G. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rauen T, Rothstein J D, Wassle H. Cell Tissue Res. 1996;286:325–336. doi: 10.1007/s004410050702. [DOI] [PubMed] [Google Scholar]
- 18.Rothstein J D, Martin L, Levey A I, Dykes Hoberg M, Jin L, Wu D, Nash N, Kuncl R W. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 19.Kanai Y, Stelzner M, Nussberger S, Khawaja S, Hebert S C, Smith C P, Hediger M A. J Biol Chem. 1994;269:20599–20606. [PubMed] [Google Scholar]
- 20.Wadiche J I, Kavanaugh M P. J Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbour B, Brew H, Attwell D. Nature (London) 1988;335:433–435. doi: 10.1038/335433a0. [DOI] [PubMed] [Google Scholar]
- 22.Kanner B I, Bendahan A. Biochemistry. 1982;21:6327–6330. doi: 10.1021/bi00267a044. [DOI] [PubMed] [Google Scholar]
- 23.Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- 24.Kanai Y, Nussberger S, Romero M F, Boron W F, Hebert S C, Hediger M A. J Biol Chem. 1995;270:16561–16568. doi: 10.1074/jbc.270.28.16561. [DOI] [PubMed] [Google Scholar]
- 25.Wadiche J I, Arriza J L, Amara S G, Kavanaugh M P. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 26.Clements J D, Westbrook G L. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- 27.Läuger P. J Membr Biol. 1987;99:1–11. doi: 10.1007/BF01870617. [DOI] [PubMed] [Google Scholar]
- 28.Eigen M, Maass G. Z Phys Chem. 1966;49:163–177. [Google Scholar]
- 29.Läuger P. J Membr Biol. 1980;57:163–178. doi: 10.1007/BF01869585. [DOI] [PubMed] [Google Scholar]
- 30.Otis T S, Kavanaugh M P. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]