

Abstract
At early developmental stages, silent synapses have been commonly found in different brain areas. These synapses are called silent because they do not respond at rest but are functional at positive membrane potentials. A widely accepted interpretation is that N-methyl-d-aspartate (NMDA) but not α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are functionally expressed on the subsynaptic membrane. Here we show that, in both CA3 and CA1 hippocampal regions, AMPA-mediated synaptic responses can be detected already at early stages of postnatal development. However, some synapses appear silent because of a very low probability of glutamate release. They can be converted into functional ones by factors that enhance release probability such as paired-pulse stimulation, increasing the temperature or cyclothiazide (CTZ), a drug that blocks AMPA receptor desensitization and increases transmitter release. Conversely, conducting synapses can be switched off by increasing the frequency of stimulation. Although we cannot exclude that “latent AMPA receptors” can become functional after activity-dependent processes, our results clearly indicate that, in the neonatal hippocampus, a proportion of glutamatergic synaptic connections are presynaptically rather than postsynaptically silent.
Silent synapses are synapses that are not conducting at rest either because neurotransmitter is not released when the presynaptic terminal is invaded by an action potential or because they are unable to detect the release of neurotransmitter (1). Their role in development and neuronal plasticity was suggested long ago (2–4). More recently, synapses that are silent at rest but functional at positive membrane potentials have attracted special attention (5–7). The most common interpretation is that these synapses do not express functional α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors but only N-methyl-d-aspartate (NMDA) receptors, which at rest are inactive because of the strong voltage-dependent magnesium block (8). It has been proposed that activity-dependent processes, such as long-term potentiation (LTP), are associated with the conversion of silent synapses into functional ones and require either insertion of new AMPA receptor proteins in the subsynaptic membrane or activation of nonfunctional receptors (5–7, 9). Although this hypothesis has been widely accepted and has been recently validated at the ultrastructural level (10, 11), alternative mechanisms can be put forward. Silent synapses can be presynaptically silent, i.e., they might release much less glutamate than functional synapses or no glutamate at all. In this case, an enhanced release of glutamate during LTP may involve an increase in the number of quanta released per presynaptic action potential. Thus, mossy fiber (MF) LTP, whose locus of expression is mainly presynaptic (ref. 12, but see ref. 13), and the late phase of CA3-CA1 LTP, which requires cAMP-dependent protein synthesis, are associated with an increase in the number of quanta released per action potential (14–16).
Silent synapses represent a common feature during postnatal development and have been observed in a variety of different structures including the hippocampus (7, 9, 17–22), where their conversion into functional ones is thought to be crucial for stabilization of synaptic contacts and for refinement of neuronal networks. In particular, in the CA1 region of the hippocampus, silent synapses are believed to represent the majority of glutamatergic synapses at early developmental stages, but no information is available regarding the CA3 region. This hippocampal area appears to be particularly interesting because, among afferent fibers to CA3 pyramidal cells, MFs have a postnatal development. In fact, although immature axons may contact pyramidal cells from the first postnatal day, full development of characteristic thorny excrescencies is reached only by the end of the second postnatal week (23).
The aim of the present work was to study whether, at the mossy fiber-CA3 synapses, glutamatergic AMPA receptors are present and functional from early postnatal days. As in the CA1 neurons, we have found that a percentage of synapses were silent at rest. However, both CA1 and CA3 connections were presynaptically silent because they could be converted into functional ones by factors that enhance release probability, such as paired-pulse stimulation, increase of temperature, or cyclothiazide (CTZ), a drug that blocks AMPA receptor desensitization and increases transmitter release.
Methods
Slices Preparation.
Experiments were performed on hippocampal slices obtained from postnatal day (P) 2–P7 Wistar rats as previously described (24). Briefly, animals were decapitated after being anesthetized with i.p. injection of urethane (2 g/kg). The brain was quickly removed from the skull and placed in an ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) 130 NaCl, 3.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 1.3 MgCl2, 2 CaCl2, and 11 glucose, and saturated with 95% O2 and 5% CO2 (pH 7.3–7.4). Transverse hippocampal slices (300–400 μm thick) were cut with a vibratome and stored at room temperature in a holding bath containing the same saline solution as above. After a recovery period of at least 1 h, an individual slice was transferred to the recording chamber where it was continuously superfused with oxygenated ACSF at a rate of 2–3 ml/min.
Electrophysiological Recordings.
Excitatory postsynaptic currents (EPSCs) from single CA1 or CA3 pyramidal neurons were recorded at 24°C or at 32°C by using the patch clamp technique in whole cell configuration. Patch pipettes were filled with a solution containing (in mM) 125 cesium methanesulphonate, 10 CsCl, 10 Hepes, 0.6–2 EGTA, 5 N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314, Alomone Labs, Jerusalem, Israel), 2 MgATP, and 0.3 NaGTP (resistance 3–5 MΩ). Bicuculline methoiodide (10 μM, Sigma) and tetrodotoxin (TTX, 10 nM, Affiniti Research Products, Exeter, U.K.) were added to the bathing solution to block γ-aminobutyric acid type A (GABAA) receptor and reduce polysynaptic activity, respectively. 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP, Tocris Cookson, Bristol, U.K.), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Tocris Cookson), and CTZ (Lilly Research Laboratories, Indianapolis) were added to the bathing solution when needed. Bipolar twisted NiCr-insulated electrodes or glass pipettes were placed in the stratum radiatum or in the stratum lucidum, to activate Schaffer collaterals or MFs, respectively. They were moved around the cell until a response was found. Paired stimuli (50-ms interval, 100-μs duration) were usually applied at 0.05 Hz. The minimal intensity necessary to evoke a response at −60 mV was found and then slightly decreased to obtain only failures. If no response could be detected at +40 mV, the stimulation intensity was slightly increased until a response was obtained at positive potentials. Similarly to a recently published paper (25), inputs were considered to be silent when 20–60 consecutive failures of AMPA responses were detected at −60 mV whereas for the same input outward NMDA-mediated synaptic currents were recorded at positive potentials. MF-EPSCs were characterized by their fast rise time (13) (1.4 ± 0.5 ms, n = 45, measured between 10% and 90% of their amplitude) and by their sensitivity to (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV, 1 μM, Tocris Cookson), a selective agonist for metabotropic glutamate receptors (mGluRs) 2/3, known to selectively block MF transmission (26). In comparison with previous data obtained from older animals (24), DCG-IV was able to significantly (P < 0.001, Wilcoxon test) reduce MF-EPSCs only in 13 of 21 cells tested. This weak effect could be attributed to an incomplete expression of mGluRs on presynaptic terminals at early postnatal stages or to deficient transduction mechanisms. Failures (N0) were usually estimated by visual discrimination. In a set of experiments to control the adequacy of the visual selection, N0 were calculated also by doubling the fraction of responses with amplitude >0 pA. A similarity and a high correlation between N0 estimated by the two respective methods (mean failure ratios were 85.4% ± 3.6% and 83.2% ± 4.6%, n = 28, r = 0.92, P < 0.0001) were found.
For each cell treated with CTZ, amplitude, rise, and decay time constants were evaluated on the average of successes at both 24°C and 32°C. To see whether CTZ has a presynaptic site of action, paired-pulse facilitation (PPF) was measured before and during CTZ application. PPF was calculated as the ratio between the amplitude of the second and first response after averaging all of the traces. In these experiments, stimulation was set at such an intensity to prevent an excessive number of failures to the first stimulus that would make PPF equal to infinity.
To see whether temperature or CTZ can affect presynaptic excitation, we recorded field excitatory postsynaptic potentials (fEPSPs) with a glass microelectrode filled with NaCl (2 M) placed in the stratum radiatum of the CA1 area. fEPSPs were evoked by pair of stimuli (50-ms interval, 0.05 Hz frequency, 100 μs duration) delivered to the Schaffer collaterals with bipolar twisted NiCr-insulated electrodes. Input-output curves were constructed by plotting the amplitude of the afferent volley, normalized to their maximum values vs. stimulation intensity, expressed as a fractional increase over threshold.
Data are presented as mean ± SEM. Statistical comparisons were made with the use of paired t test or Wilcoxon signed rank test (P < 0.05 was taken as significant).
Results
Most experiments on silent synapses have been done at room temperature (20°C–25°C). Our first set of experiments was done instead at 32°C. We used the whole-cell configuration of the patch clamp technique to record CA3 pyramidal neurons in hippocampal slices from rats at postnatal day P2 to P7. EPSCs were elicited by minimal stimulation of the MF. In Fig. 1a, when the cell was held near its resting potential (−60 mV), no synaptic responses could be recorded to the first stimulus. However, occasional (8/25) EPSCs, fluctuating in amplitude from trial to trial, were seen after the second stimulus. These EPSCs were completely blocked by CNQX (10 μM, not shown), implying that they were generated by glutamate acting on non-NMDA receptors. When held at +40 mV, the cell showed EPSCs with slow kinetics after both the first and second stimulus (Fig. 1a Middle). The contribution of a NMDA-mediated component to the EPSCs was ascertained by applying the selective NMDA receptor antagonist CPP (20 μM). As shown in Fig. 1a (Bottom), CPP revealed a fast AMPA-mediated component. Thus, it appears that this synapse was “silent” to the first stimulus, not because of the absence of AMPA receptors, but rather because of the low probability of glutamate released. Overall, only 2 of 29 cells were silent to the first stimulus under our experimental conditions (32°C, 0.05 Hz testing frequency). On average, the percentage of successes to the first stimulus at −60 mV was 34.2% ± 3.8%. When held at +40 mV, all neurons showed slow kinetics EPSCs with significantly higher percentage of successes (55.9% ± 4.9%, P < 0.001, Wilcoxon test; Fig. 1c). The presence of AMPA-mediated EPSCs after the second stimulus at −60 mV and +40 mV allows us to conclude that, at 32°C, postsynaptic AMPA receptors can be revealed at MF-CA3 synapses even at early developmental stages.
Figure 1.
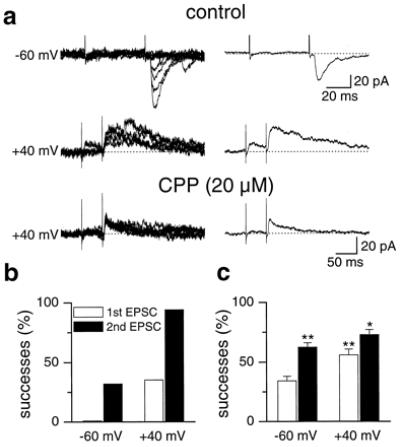
Neonatal MF-CA3 synapses, silent to the first stimulus, respond to the second in a paired-pulse paradigm. (a) Eight to ten individual traces (Left) obtained at 32°C by pair of stimuli (50-ms interval) delivered to the MF (frequency: 0.05 Hz) at−60 mV and at +40 mV in control conditions and in the presence of CPP (P5 rat). Averages of successes to the second stimulus (Top and Bottom) or to the first stimulus (Middle) are shown on the Right. (b) Percentage of successes to the first (open column) and to the second stimulus (filled column) at −60 and +40 mV for the neuron shown in a. (c) Mean percentage of successes to the first and to the second stimulus at −60 and +40 mV for all P2-P7 CA3 pyramidal neurons examined at 32°C (n = 29). At −60 mV, the percentage of successes to the second stimulus is significantly higher than that to the first one, whereas, at +40 mV, the values are significantly higher than those at −60 mV for both the stimuli (*, P < 0.05; **, P < 0.001; Wilcoxon test).
It is known that the reliability of synaptic transmission decreases with decreasing temperature, apparently because of reduced transmitter release probability (27). To examine this effect, EPSCs were recorded first at 24°C and then at 32°C. At 24°C, in two of 12 cells held at −60 mV, responses to the first stimulus were absent whereas responses to the second one could be rarely recorded (in the cell shown in Fig. 2a, the second stimulus induced 3 responses of 18 trials). When the temperature was raised to 32°C, EPSCs to the first stimulus appeared, and the number of responses to the second was clearly enhanced (14/32; Fig. 2a). On average in 7 cells, increasing the temperature from 24°C to 32°C produced a significant increase in the percentage of successes to the first stimulus (from 9.3% ± 3.5% to 20.9% ± 4.5%, P < 0.05, Wilcoxon test; Fig. 2b). Thus, synaptic activity in the CA3 hippocampal region is strongly temperature-dependent.
Figure 2.
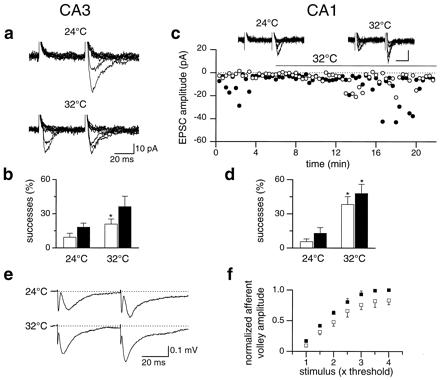
Temperature dependency of MF-CA3 and Schaffer collaterals-CA1 synaptic transmission. (a) Paired-pulse stimulation of MF at 24°C evoked no responses to the first stimulus but some to the second one (P4 rat). Raising the temperature from 24°C to 32°C resulted in the appearance of responses to the first stimulus and increased the number of successes to the second (ten traces are shown for both conditions). (b) Mean percentage of successes to the first and to the second stimulus at 24°C and 32°C for all P2-P7 CA3 pyramidal neurons examined (n = 7). The percentage of successes to the first stimulus at 32°C was significantly higher than at 24°C (P < 0.05, Wilcoxon test). (c) Peak amplitude of the first (open circles) and second (filled circles) EPSCs recorded from the CA1 region at 24°C and at 32°C (bars) plotted against time. (Insets) Five individual traces of EPSCs evoked by paired stimulation of Schaffer collaterals at 24°C and 32°C. Calibration bars: vertical, 20 pA; horizontal, 20 ms (P2 rat). Note absence of responses to the first stimulus at 24°C. Increasing the temperature from 24°C to 32°C resulted in the appearance of some responses to the first stimulus and enhanced the number of successes to the second. (d) Mean percentage of successes to the first and to the second stimulus at 24°C and 32°C for all P2-P6 CA1 pyramidal neurons tested (n = 8). The values at 32°C are significantly higher for both the first and the second EPSC (P < 0.01, Wilcoxon test). (e) Pair of fEPSPs (average of three traces) evoked at 0.05 Hz by stimulation of Schaffer collaterals at 24°C and 32°C from a P6 rat. (f) Amplitude of the afferent volley, normalized to the maximum amplitude vs. stimulus strength, expressed as X times the threshold at 24°C (filled squares) and 32°C (open squares). Each symbol represents the mean of four experiments. Bars are SEM.
A similar temperature effect was seen when paired-pulse minimal stimulation of Schaffer collaterals was used to induce EPSCs in CA1 pyramidal neurons in P2–P4 hippocampal slices (Fig. 2 c and d). At 24°C, 10 of 28 CA1 neurons exhibited no successes to the first stimulus, but 7 of them showed sporadic responses to the second. However, as for MF input, NMDA-receptor-mediated EPSCs could be detected at +40 mV (not shown). It should be stressed that, at 24°C, the probability of successes to the first stimulus was extremely low even in those synapses that were not silent, sometimes only a single response being detected in more than 20 trials. As shown in the example of Fig. 2c, raising the temperature from 24°C to 32°C resulted in the appearance of AMPA-mediated responses to the first stimulus and an increase in the number of second responses (from 6/20 to 12/26). On average, in 8 of 10 cells recorded at both 24°C and 32°C, the percentage of successes to the first and second stimulus increased significantly (P < 0.01, Wilcoxon test) from 5.6% ± 2.4% and 12.9% ± 5.0% to 38.2% ± 6.9% and 47.9% ± 8.1%, respectively (Fig. 2d).
These results suggest that the absence of synaptic responses in both MF-CA3 and Schaffer collateral-CA1 synapses in early postnatal life is not due to the absence of functional AMPA receptors, but rather to a low level of glutamate release, particularly at room temperature. According to this hypothesis, the conversion of silent synapses into functional ones, observed after raising the temperature from 24°C to 32°C, could be due to an increase in neurotransmitter release. However, it cannot be excluded that this procedure induces a change in presynaptic cell excitability, so that the same stimulus may recruit more presynaptic fibers. To test this hypothesis, possible changes in the amplitude of extracellular fiber volley (an index of presynaptic excitation) were monitored by recording fEPSPs evoked in the CA1 region by pair of stimuli. As shown in Fig. 2e, the amplitude of the afferent volley preceding the first and second response was exactly the same. Increasing the temperature from 24°C to 32°C, caused an increase in the amplitude of the fEPSP (from 139 ± 32 to 212 ± 47 μV, at 2–2.5 times the threshold, n = 4) and a reduction in the peak latency (from 8.25 ± 0.54 to 5.62 ± 0.13 ms, see Fig. 2e). Moreover, as shown in Fig. 2 e and f, a small but consistent reduction in the amplitude of the afferent volley was observed. At 2.5 times the threshold, the normalized amplitude changed from 0.81 ± 0.06 at 24°C, to 0.66 ± 0.10 at 32°C (n = 4). It is therefore likely that temperature-dependent conversion of silent synapses into functional ones is not due to recruitment of new afferent fibers but requires an increase in neurotransmitter release. To further examine this possibility, we applied CTZ, a drug known to potentiate glutamate release and to selectively block AMPA receptor desensitization (refs. 28 and 29, but see also ref. 25). Before CTZ application, 4 out of 10 cells were silent. During CTZ application (50 μM), two of four silent synapses became conductive (Fig. 3 a and b). In another six cells, CTZ induced an increase in the percentage of successes to the first stimulus (from 0.31% ± 0.08% to 0.45% ± 0.08%) and a significant increase in the amplitude of the first response (from −25.9 ± 8.4 to −33.3 ± 7.9 pA, P < 0.05, Wilcoxon rank test). Moreover, in agreement with previous studies (25, 30), CTZ enhanced the rise time and the decay time constant. For the first response, the rise time varied from 1.23 ± 0.21 to 2.06 ± 0.19 ms (n = 5, P < 0.05, paired t test) and the decay time constant from 7.1 ± 1.2 to 15.4 ± 1.8 ms, (n = 5, P < 0.005, paired t test, Fig. 3a). To see whether the potentiating effect of CTZ on minimal EPSCs was due to a presynaptic site of action, the PPF of synaptic responses elicited by stimulation of the Schaffer collaterals was measured before and during CTZ perfusion. CTZ reduced PPF from 2.04 ± 0.15 to 1.35 ± 0.13 (P < 0.01, paired t test, n = 6). In extracellular recordings, CTZ did not increase the amplitude of the afferent volley preceding the fEPSP (at 2.5 times the threshold, the normalized amplitude was 0.76 ± 0.04 before and 0.69 ± 0.05 during CTZ application, n = 4). From these experiments, we can conclude that CTZ increases transmitter release without recruiting more presynaptic fibers.
Figure 3.
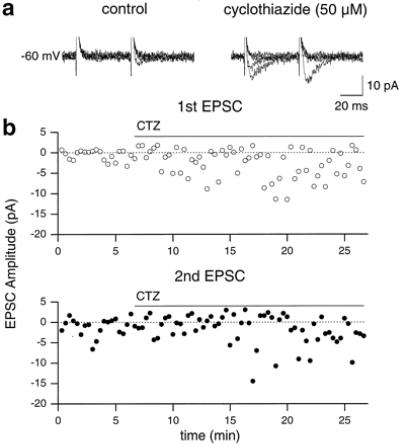
Effect of CTZ on synaptic responses at Schaffer collaterals-CA1 synapses. (a) EPSCs recorded at 24°C (P3 rat). Bath perfusion of CTZ (50 μM, bars) induced the appearance of successes to the first stimulus (in both conditions, five traces are shown). (b) Time course of CTZ effect on the amplitude of the first (open circles) and second (filled circles) EPSCs, before, during, and after CTZ application (50 μM, bars) for the cell shown in a.
In previous studies, silent synapses were considered silent if they did not exhibit any AMPA-mediated responses in 25–100 consecutive trials (at stimulation frequencies of 0.3–2 Hz; refs. 5, 6, and 25). To see whether our results were consistent with previous published papers, additional experiments were performed using stricter criteria (at the stimulation frequency of 0.5 Hz). Synapses were considered “silent” when they showed no responses to the first stimulus in >60 consecutive trials. The chance of observing >60 failures in a row would be <5% for a release probability <0.05. Of 13 tested cells, 7 (53.8%) were silent to the first stimulus in 60–70 consecutive trials but showed rare responses to the second one (on average 8.0% ± 2.6%). Among seven cells recorded at both 24°C and 32°C, four became conductive to the first stimulus at higher temperature whereas, in the remaining three, the percentage of successes to both stimuli increased considerably. On average, the mean percentage of successes to the first and second stimulus was significantly enhanced (from 1.6% ± 0.9% and 4.2% ± 1.0% at 24°C to 25.6% ± 9.5% and 27.0% ± 6.9% at 32°C, respectively; n = 7, P < 0.05, Wilcoxon test; Fig. 4 a and b). These results indicate that synapses identified as “silent” with more strict criteria can be switched on by factors that enhance release probability.
Figure 4.
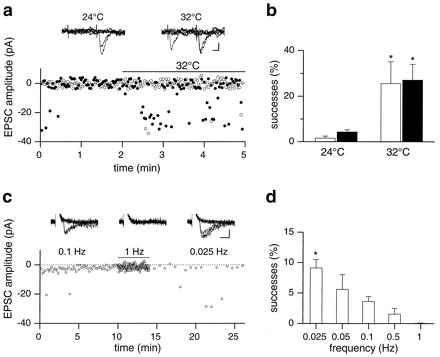
Effect of temperature (a and b) and stimulus frequency (c and d) on EPSCs elicited in CA1 pyramidal neurons by Schaffer collateral stimulation. (a) PPS (at 0.5 Hz) of Schaffer collaterals evoked no responses to the first stimulus (open circles) but some to the second one (filled circles; P5 rat) at 24°C. Raising the temperature from 24°C to 32°C resulted in the appearance of responses to the first stimulus and increased the number of successes to the second. Insets represent six individual traces recorded at 24°C and 32°C, respectively (calibration bars: vertical, 10 pA; horizontal, 10 ms). (b) Mean percentage of successes to the first (open column) and to the second (filled column) stimulus at 24°C and 32°C (n = 7). At 32°C, the percentage of successes to both stimuli was significantly higher than at 24°C (P < 0.05, Wilcoxon test). (c) Effects of frequency changes in a representative experiment. Stimulation frequency was changed from 0.1 to 1 Hz (bar) and then to 0.025 Hz. Notice the disappearance of the EPSC during 0.1 Hz stimulation. (Insets) Seven individual traces of EPSCs evoked at 0.1, 1, and 0.025 Hz. Calibration bars: vertical, 15 pA; horizontal, 5 ms. (d) Mean percentage of successes obtained in eight cells by changing the stimulation frequencies as in a. For comparison, the mean percentage of successes obtained at 0.5 Hz (n = 7 cells) and 0.05 Hz stimulation (n = 8) in two additional sets of experiments is shown. The value obtained at 0.025 Hz is significantly different from that at 0.1, 0.5, or 1 Hz (P < 0.05 and P < 0.01, respectively, Wilcoxon test). Note that synapses showed a low efficacy at all frequencies and became silent during 1-Hz test stimulation.
Despite the low frequency stimulation used in the majority of our experiments (0.05 Hz), EPSCs tended to disappear with time (see the sequence of second EPSC in Fig. 2c). This synaptic depression, which occurred in the absence of any change in leak conductance of the cell, could be reversed by increasing the temperature or by slowing down the rate of stimulation. To see whether increasing stimulation frequency could convert functional synapses into silent ones, Schaffer collaterals were stimulated first at 0.1 Hz and then at 1 Hz. At 0.1 Hz, the percentage of successes was very low, leading to silent synapses in two of eight cells. During 1 Hz stimulation, all synapses became silent so that in >60 trials only failures to both stimuli were recorded (Fig. 4 c and d). In all neurons tested, EPSCs reappeared after reducing the stimulation frequency from 1 to 0.025 Hz. These results indicate that, in early postnatal life, synaptic reliability strongly depends on stimulation rate and that, at relatively modest frequencies of stimulation, conducting synapses can be converted into silent ones.
Discussion
In this study, we have shown that both MF-CA3 and Schaffer collateral-CA1 synapses express functional AMPA receptors from the first postnatal days, but they may appear silent at resting membrane potential because of a very low probability of glutamate release. Several lines of evidence suggest that these synapses are presynaptically silent. (i) They could respond to a second stimulus in a paired-pulse protocol. PPF is a widely accepted index of presynaptic function (31) and was observed also in those cases in which rare successes to the first stimulus were detected. It should be stressed that the paired-pulse stimulation procedure mimics repetitive firing, known to play a crucial role in information processing, thus making synaptic transmission more reliable (32). (ii) These synapses could become conductive when the temperature was raised from 24°C to 32°C. Moreover, synapses already functional at 24°C showed an enhanced percentage of successes at 32°C. Temperature is known mainly to affect processes involved in synaptic release, for example, increasing the mean number of transmitter quanta released per presynaptic action potential (27). The possibility that temperature affects presynaptic cell excitability so that, at higher temperature, the same stimulus may recruit more fibers can be ruled out. In extracellular experiments, raising the temperature from 24°C to 32°C did not produce any significant increase in the amplitude of the afferent volley; a tendency toward a reduction was instead constantly observed. These data are in agreement with a previous study in which rising temperature caused a reduction in the duration of the directly evoked action potential in hippocampal slices from immature rabbits (33). (iii) These synapses could become functional in the presence of CTZ, which is known to block AMPA receptor desensitization and to enhance glutamate release (refs. 28 and 29, but see ref. 25). In line with an increase in release probability is the observation that CTZ not only considerably enhanced the percentage of successes to the first stimulus but also significantly decreased the PPF ratio. These results are in agreement with recent findings suggesting that, in the immature hippocampus, silent synapses containing an appreciable number of AMPA receptors appear silent because the peak of glutamate concentration in the synaptic cleft is too low to activate low-affinity AMPA receptors (25). Low levels of glutamate could arise either from spillover of neurotransmitter from neighboring terminals (34–36) and/or by the trickle of neurotransmitter from a fusion pore as occurs in neuronal (37) or non-neuronal secretory cells (38–40). According to the spillover hypothesis, only high-affinity NMDA receptors, but not low-affinity AMPA receptors, would be activated by the low concentration of glutamate diffused from a neighboring synapse (35, 41). An enhanced probability of neurotransmitter release—after PPF, increased temperature, and CTZ—would allow glutamate to be released also locally close to the subsynaptic membrane so that its concentration would be high enough to activate both NMDA and AMPA receptors, thus converting silent synapses into functional ones. The higher percentage of silent synapses at 24°C rather than at 32°C, found in the present experiments, is also in favor of the spillover hypothesis (34, 35). However, at physiological temperature, spillover would be limited by the higher rate of glutamate clearance from the extracellular space (34, 36), and, therefore, the contribution of this mechanism should be minimal. Alternatively, release of a small amount of glutamate from a nonexpanding fusion pore may account for some of the above observations (37–40). Factors that increase the release probability would modify the gating properties of presynaptic fusion pores from the nonexpanding mode to a rapidly expanding one in the same way as recently suggested for LTP at silent synapses (25).
Additional evidence in favor of a presynaptic mechanism is given by the experiments performed at different stimulation frequencies. The short-term synaptic depression and the appearance of silent synapses induced by increasing stimulation rate are most likely attributable to changes in presynaptic release mechanisms. Using multiple-probability fluctuation analysis at rat climbing fiber-Purkinje cell synapses, it was found that frequency-dependent depression was associated with a dramatic fall in release probability, with no apparent changes in the number of functional release sites or in the quantal size, suggesting a pure presynaptic origin (42). At the Schaffer collateral-CA1 synapse of the adult hippocampus, a reduction in quantal content because of changes in release probability was found during low-frequency depression, although it was sometimes accompanied by changes in the quantal size (43). The slow reappearance of responses observed in the present experiments may involve recovery from presynaptic calcium channel inactivation (44) or distinct kinetic phases in vesicle recycling (45).
Altogether, the above results strengthen the role of presynaptic mechanisms in controlling synaptic efficacy in the developing hippocampus. In comparison with previous work (7, 9), in the present experiments the percentage of Schaffer collateral-CA1 synapses silent at rest was smaller. This apparent discrepancy could be attributed to differences in recording conditions. In particular, previously reported data were obtained at higher frequency of stimulation (0.1–0.3 Hz), conditions that, according to our results, would favor the reduction in the percentage of successes and eventually would lead to the disappearance of synaptic responses. In keeping with this prediction, also in our experiments the percentage of silent synapses increased to 53.8% when the stimulation frequency was 0.5 Hz.
Although the above results do not preclude the possibility that functional AMPA receptors can be recruited at postsynaptic sites during LTP (10, 11, 46, 47), they strengthen the role of presynaptic mechanisms in controlling synaptic efficacy in a proportion of hippocampal neurons during early postnatal development.
Acknowledgments
We thank Prof. A. R. Martin and J. G. Nicholls for helpful discussion and carefully reading the manuscript. This work was supported by grants from Ministero Dell'Università e Ricerca Scientifica (MURST) to E.C. and from the International Association for the Promotion of Co-operation with Scientists from the New Independent States of the Former Soviet Union (INTAS) to E.C. and L.L.V.
Abbreviations
- NMDA
N-methyl-d-aspartate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CTZ
cyclothiazide
- MF
mossy fibers
- EPSC
excitatory postsynaptic current
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CPP
3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid
- LTP
long-term potentiation
- PPF
paired-pulse facilitation
- fEPSP
field excitatory postsynaptic potential
- Pn
postnatal day n
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170032297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170032297
References
- 1.Malenka R C, Nicoll R A. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- 2.Jack J J, Redman S J, Wong K. J Physiol (London) 1981;321:111–126. doi: 10.1113/jphysiol.1981.sp013974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voronin L L. Neuroscience. 1983;10:1051–1069. doi: 10.1016/0306-4522(83)90099-4. [DOI] [PubMed] [Google Scholar]
- 4.Faber D S, Lin J W, Korn H. Ann N Y Acad Sci. 1991;627:151–164. doi: 10.1111/j.1749-6632.1991.tb25920.x. [DOI] [PubMed] [Google Scholar]
- 5.Liao D, Hessler N A, Malinow R. Nature (London) 1995;455:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 6.Isaac J T R, Nicoll R A, Malenka R C. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 7.Durand G M, Kovalchuk Y, Konnerth A. Nature (London) 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- 8.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Nature (London) 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 9.Liao D, Malinow R. Learn Mem. 1996;3:138–149. doi: 10.1101/lm.3.2-3.138. [DOI] [PubMed] [Google Scholar]
- 10.Shi S H, Hayashi Y, Petralia R S, Zaman S H, Wenthold R J, Svoboda K, Malinow R. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 11.Liao D, Zhang X, O'Brien R, Ehlers M D, Huganir R L. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- 12.Zalutsky R A, Nicoll R A. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 13.Yeckel M F, Kapur A, Johnston D. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tong G, Malenka R C, Nicoll R A. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- 15.Bolshakov V Y, Golan H, Kandel E R, Siegelbaum S A. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 16.Ma L, Zablow L, Kandel E R, Siegelbaum S A. Nat Neurosci. 1999;2:24–30. doi: 10.1038/4525. [DOI] [PubMed] [Google Scholar]
- 17.Wu G-Y, Malinow R, Cline H T. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Wojtowicz J M, Atwood H L. Synapse. 1996;22:78–86. doi: 10.1002/(SICI)1098-2396(199601)22:1<78::AID-SYN9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 19.Isaac J T R, Crair M C, Nicoll R A, Malenka R C. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Zhuo M. Nature (London) 1998;393:695–698. doi: 10.1038/31496. [DOI] [PubMed] [Google Scholar]
- 21.Bardoni R, Magherini P C, MacDermott A B. J Neurosci. 1998;18:6558–6567. doi: 10.1523/JNEUROSCI.18-16-06558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rumpel S, Hatt H, Gottmann K. J Neurosci. 1998;18:8863–8874. doi: 10.1523/JNEUROSCI.18-21-08863.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amaral D G, Dent J A. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- 24.Domenici M R, Berretta N, Cherubini E. Proc Natl Acad Sci USA. 1998;95:8310–8315. doi: 10.1073/pnas.95.14.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi S, Klingauf J, Tsien R W. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- 26.Kamiya H, Shinozaki H, Yamamoto C. J Physiol (London) 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardingham N R, Larkman A U. J Physiol (London) 1998;507:249–256. doi: 10.1111/j.1469-7793.1998.249bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diamond J S, Jahr C E. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 29.Bellingham M C, Walmsley B. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 30.Rammes G, Zeilhofer H U, Collingridge G L, Parsons C G, Swandulla D. Pflügers Arch. 1999;437:191–196. doi: 10.1007/s004240050768. [DOI] [PubMed] [Google Scholar]
- 31.Zucker R S. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- 32.Lisman J E. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- 33.Shen K, Schwartzkroin P A. Brain Res. 1988;20:305–316. doi: 10.1016/0006-8993(88)90619-1. [DOI] [PubMed] [Google Scholar]
- 34.Asztely F, Erdemli G, Kullmann D M. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 35.Kullmann D M, Asztely F. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- 36.Min M Y, Rusakov D A, Kullmann D M. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 37.Bruns D, Jahn R. Nature (London) 1995;377:62–65. doi: 10.1038/377062a0. [DOI] [PubMed] [Google Scholar]
- 38.Spruce, A. E., Breckenridge, L. J., Lee, A. K. & Almers, W. Neuron4, 643–654. [DOI] [PubMed]
- 39.Zhou Z, Milser S, Chow R H. Biophys J. 1996;70:1543–1552. doi: 10.1016/S0006-3495(96)79718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lollike K, Borregaard N, Lindau M. Biophys J. 1998;75:53–59. doi: 10.1016/S0006-3495(98)77494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patneau D K, Mayer M L. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silver R A, Momiyama A, Cull-Candy S G. J Physiol (London) 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larkman A U, Jack J J, Stratford K J. J Physiol (London) 1997;505:457–471. doi: 10.1111/j.1469-7793.1997.457bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gingrich K J, Byrne J H. J Neurophysiol. 1985;53:652–669. doi: 10.1152/jn.1985.53.3.652. [DOI] [PubMed] [Google Scholar]
- 45.Dittman J S, Regehr W G. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petralia R S, Esteban J A, Wang Y X, Partridge J G, Zhao H M, Wenthold R J, Malinow R. Nat Neurosci. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- 47.Malenka R C, Nicoll R A. Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]