

Abstract
VacA is a secreted toxin that plays a role in Helicobacter pylori colonization of the stomach and may contribute to the pathogenesis of peptic ulcer disease and gastric cancer. In this study, we analyzed a library of plasmids expressing randomly mutated forms of recombinant VacA and identified 10 mutant VacA proteins that lacked vacuolating cytotoxic activity when added to HeLa cells. The mutations included six single amino acid substitutions within an amino-terminal hydrophobic region and four substitutions outside the amino-terminal hydrophobic region. All 10 mutations mapped within the p33 domain of VacA. By introducing mutations into the H. pylori chromosomal vacA gene, we showed that secreted mutant toxins containing V21L, S25L, G121R, or S246L mutations bound to cells and were internalized but had defects in vacuolating activity. In planar lipid bilayer and membrane depolarization assays, VacA proteins containing V21L and S25L mutations were defective in formation of anion-selective membrane channels, whereas proteins containing G121R or S246L mutations retained channel-forming capacity. These are the first point mutations outside the amino-terminal hydrophobic region that are known to abrogate vacuolating toxin activity. In addition, these are the first examples of mutant VacA proteins that have defects in vacuolating activity despite exhibiting channel activities similar to those of wild-type VacA.
Infection by Helicobacter pylori is associated with an increased risk for development of peptic ulcer disease, gastric adenocarcinoma, and gastric lymphoma (5, 15, 45). An exotoxin produced by H. pylori, VacA, was discovered in 1988 (27) and was subsequently shown to contribute to the capacity of H. pylori to colonize the stomach in a murine model of infection (42). Substantial evidence indicates that VacA plays a role as a virulence factor in the pathogenesis of H. pylori-induced gastric diseases (6, 32, 36).
One of the most striking activities of VacA is its capacity to induce the formation of large cytoplasmic vacuoles in cultured cells (7, 27). VacA also produces an assortment of other effects, including depolarization of the cellular membrane potential, apoptosis, detachment of cells from the basement membrane, interference with the process of antigen presentation, and activation of mitogen-activated protein kinases (6, 32, 36). VacA interferes with the activation and proliferation of T cells (3, 19, 46), which might be a factor that enables H. pylori to resist clearance by host immune defenses.
VacA-induced cell vacuolation requires binding of VacA to the surface of cells, internalization of VacA into cells, and insertion of VacA into membranes to form anion-selective membrane channels (6, 18, 32). VacA channels are formed in the plasma membrane of cells and also may be present in the membranes of endocytic vesicles (47, 49). One model proposes that entry of Cl− into vesicle compartments through the VacA channel is accompanied by increased pumping of protons by the vacuolar ATPase, influx of weak bases (such as ammonium ions), and accumulation of H2O, thereby driving endosome swelling (1, 37, 49). It has been suggested that VacA mimics the electrophysiological behavior of host chloride channels (11). Indeed, overexpression of the endogenous chloride channel ClC-3 results in cytoplasmic vacuolation that is remarkably similar to the vacuolation induced by the VacA toxin (28). Several other activities attributed to VacA also are associated with the capacity of the toxin to form anion-selective membrane channels (10, 23, 33, 46, 47, 57).
The VacA primary amino acid sequence is not closely related to that of any other known bacterial toxin, and the three-dimensional structure of VacA has not yet been determined. Partial proteolytic cleavage of the 88-kDa VacA protein with trypsin yields an amino-terminal p33 fragment (amino acids 1 to 312) and a carboxy-terminal p55 fragment (amino acids 313 to 821) (8, 17, 38, 48, 51). It has been suggested that these represent two domains of VacA. Both domains are required for toxin activity (50, 52, 61, 63).
In an effort to identify regions of VacA that are required for toxin activity, inactive mutant VacA proteins have been characterized (12, 30, 39, 55, 61-63). One approach for generating inactive VacA mutant proteins has involved introduction of mutations into the chromosomal vacA gene in H. pylori. Several mutant proteins generated by this approach contain large deletions that are expected to drastically alter VacA structure (55). Most of the other previously described inactive mutant VacA proteins contain mutations in a region near the amino terminus (amino acids 1 to 32) (12, 30, 55, 62, 63), which is predicted to be highly hydrophobic and which plays a role in the formation of anion-selective channels (30, 55). The goals of the present study were to screen a library of mutant VacA toxins expressed in Escherichia coli and identify mutant proteins that lack vacuolating activity.
MATERIALS AND METHODS
Mutagenesis of vacA.
A plasmid (pMM592) expressing recombinant VacA (rVacA) (31) was transformed into mutator E. coli strain XL1-Red (Stratagene). After 48 h of growth on plates, colonies were pooled and plasmid DNA was extracted. The resulting library of mutated plasmids then was transformed into E. coli expression strain ER2566, which encodes an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible copy of the RNA polymerase gene from bacteriophage T7.
Expression of VacA proteins in E. coli.
Expression of rVacA and preparation of soluble protein extracts was performed with 96-well deep-well plates as described previously (31). ER2566 transformants were grown at 25°C overnight with shaking in Terrific broth (TB; per liter, 12 g tryptone, 24 g yeast extract, 4 ml glycerol, 2.31 g KH2PO4, 12.54 g K2HPO4) supplemented with 25 μg/ml kanamycin (TB + Kan). Overnight cultures were diluted 1:100 in fresh TB + Kan and grown at 25°C for 4 h (to an optical density at 600 nm of 0.2 to 0.4). IPTG then was added to a final concentration of 250 μM, and growth was continued for 20 h at 25°C. IPTG-induced cultures were pelleted, washed in 0.9% NaCl, and resuspended in a solution (25 μl/ml of starting culture) that contained 10 mM Tris (pH 7.5), 100 mM NaCl, 1 mM EDTA, protease inhibitors (Complete mini; Roche), and 20,000 U/ml ReadyLyse lysozyme (Epicenter). Bacteria were incubated at room temperature for 15 min with periodic mixing, after which a solution (75 μl/ml of starting culture) containing 50 mM Tris (pH 8.0), 2.67 mM MgCl2, and 67 U/ml Benzonase nuclease (Novagen) was added. Samples were mixed briefly and subjected to four successive rounds of freezing (in a dry-ice-methanol bath) and thawing at 37°C. The insoluble debris was pelleted, and the supernatants were then tested immediately in a cell culture assay.
Cell culture methodology.
Vacuolating activity in E. coli extracts was examined as described previously (31). Aliquots from each extract were added to the medium overlying HeLa cells. For these assays, HeLa cells were incubated in serum-free Eagle's medium supplemented with 10 mM ammonium chloride. Vacuolation of HeLa cells was assessed by microscopic inspection. Mutants that did not exhibit vacuolating activity based on microscopic observation were examined in more detail. All mutant proteins identified in the initial screen as lacking cytotoxic activity were immunoblotted with anti-VacA antiserum no. 958 (43) to assess levels of VacA expression and to assess the sizes of the mutant VacA proteins. Mutants expressing VacA proteins that were substantially degraded, truncated, insoluble, or expressed at markedly reduced levels relative to a wild-type control were not studied further. Mutants that expressed a form of VacA that appeared to be full length and present in a quantity similar to that of the wild-type control were retested. New cultures of these mutants were grown, toxin concentrations were normalized based on an antibody capture enzyme-linked immunosorbent assay (ELISA) (54), and equal amounts of wild-type or mutant rVacA proteins were added in triplicate to HeLa cell monolayers.
Introduction of vacA mutations into H. pylori and purification of VacA from H. pylori.
Several mutations were introduced into the chromosomal vacA gene of H. pylori 60190, either with an upstream chloramphenicol resistance cassette as a selectable marker (for V21L and S25L) or by a sacB-based mutagenesis approach (4, 30, 33, 55). H. pylori strains were grown in sulfite-free brucella broth containing activated charcoal, and mutant forms of VacA were purified in an oligomeric form from H. pylori culture supernatants (8). In brief, broth culture supernatant proteins from each mutant H. pylori strain were concentrated by precipitation with a 50% saturated solution of ammonium sulfate, and the VacA proteins from each strain were isolated by fractionation with a Superose 16/50 gel filtration column. Purified VacA preparations were routinely acid activated before the addition of VacA to cell culture wells or planar lipid bilayer chambers as described previously (8, 13).
ELISA to quantify binding of VacA to cells.
VacA proteins (2.5 μg/ml) purified from H. pylori were incubated with HeLa cell monolayers at 4°C for 1 h. Monolayers were then washed to remove unbound toxin and fixed in 50% acetone-50% methanol. Bound VacA was then detected by ELISA, with rabbit anti-VacA serum no. 958 and horseradish peroxidase-labeled anti-rabbit immunoglobulin G (43).
Confocal-microscopy methodology.
Internalization of VacA mutant proteins into HeLa cells was analyzed by confocal microscopy with anti-VacA serum no. 958 as described previously (43, 50).
Analysis of protein dimerization with a TOXCAT model system.
The TOXCAT system was developed by Russ and Engelman to study transmembrane helix-helix associations in a natural membrane environment (41). In this system, a putative transmembrane sequence (TM) is cloned between a sequence encoding the transcription activator domain of Vibrio cholerae ToxR and a sequence encoding the periplasmic domain of the Escherichia coli maltose binding protein (MBP). Dimerization of the fusion protein is determined based on expression of the cat gene, which is under the control of the dimerization-dependent transcription activator ToxR (13). E. coli strains expressing ToxR-TM-MBP fusion proteins that dimerize are resistant to chloramphenicol, whereas strains expressing fusion proteins that lack a dimerization sequence are sensitive to chloramphenicol. The chloramphenicol acetyltransferase (CAT) enzyme was measured by an antigen capture ELISA (Roche) according to the manufacturer's instructions. Plasmids pccVacA-wt and pccVacA-G14A have been described previously (29), and plasmids containing additional mutations in VacA were constructed by a similar approach.
Planar lipid bilayer methodology.
Planar lipid bilayers composed of egg phosphatidylcholine-dioleoylphosphatidylserine-cholesterol (55:15:30 mol%) dissolved in n-decane, were prepared as described previously (10, 30, 33, 55). Purified, acid-activated VacA proteins (30 nM, ∼2.7 μg/ml) were added to the lipid bilayers in buffers as described in Table 1. The time required to produce a current of 100 pA at −50 mV was then determined. Membrane currents were measured as described previously (10, 30, 33, 55). The potential is indicated relative to the cis side, defined as the chamber to which the protein was added. Permeability ratios were determined from the Goldman-Hodgkin-Katz equation, after the membrane voltage for zero current (reversal potential) in asymmetric salt concentrations was measured.
TABLE 1.
Channel-forming properties of wild-type and mutant VacA proteins
| Protein | Time to −100 pA at −50 mV (min)a | Selectivityb | Ratio of avg currents (+50 mV/−50 mV)c | Ratio of channel formation rates (+50 mV/−50 mV)c |
|---|---|---|---|---|
| Wild type | 33 ± 12 (9) | 7.3 ± 1.4 (14) | 1.3 ± 0.1 (16) | 0.76 ± 0.33 (20) |
| V21L mutant | >160d (4) | NDe | ND | ND |
| V25L mutant | >615d (4) | ND | ND | ND |
| G121R mutant | 29 ± 13 (5) | 6.8 ± 0.8 (6) | 1.3 ± 0.1 (14) | 4.7 ± 1.9 (18) |
| S246L mutant | 38 ± 14 (6) | 6.9 ± 0.7 (6) | 1.3 ± 0.1 (13) | 2.8 ± 1.2 (18) |
VacA preparations(30 nM, ∼2.7 μg/ml) were added to planar lipid bilayers in a buffer containing 5 mM citric acid, 100 mM sodium chloride, and 2 mM EDTA (pH 4). The time required to produce a current of 100 pA at −50 mV was then determined. Results represent the mean ± standard deviation from multiple independent determinations for each sample tested. The number of successfully completed experiments for each sample is in parentheses.
Ion selectivity ratios (PCl/PNa) are shown. Reversal potential was measured in cis (200 mM NaCl) and in trans (100 mM NaCl) with VacA samples at 30 nM. The number of experiments is in parentheses. Results represent means ± standard deviations.
The number of measurements obtained from three separate experiments is in parentheses.
After prolonged incubation with these toxins, the membrane frequently ruptured before any stable current was observed. The average time at which the membrane ruptured is indicated.
ND, not determined.
The ratio of the channel formation rates at different voltage polarities was determined by switching the voltage back and forth between +50mV and −50mV and comparing the current preceding a change in polarity with the current after the jump back to the original polarity. These experiments were performed under conditions in which the increase in current at either polarity was linear. Therefore, if there is no difference in the rate of channel formation at the two potentials, the magnitude of the current after the jump back should be the same as that determined by simply extrapolating the linear current rise before the jump. A greater rate of channel formation during the sojourn at the other polarity would cause the current magnitude after the jump back to be greater than the extrapolated value, whereas a lower rate during the sojourn would result in a lower current magnitude after the jump back than the extrapolated value. A measurement of the difference in channel forming rates at the two potentials can be obtained as follows. The macroscopic current magnitude, I, is determined by I = N · I0, where N is the number of channels and I0 is the average current per channel. If there are N1 channels before the jump and the rate of channel formation at this potential is R1, then the extrapolated current magnitude after the jump is Iex = (N1 + R1 · t) · I0, where the duration of the sojourn is t. If the channel formation rate at the other potential is R2, then the actual current magnitude after the jump is Iact = (N1 + R2 · t) · I0. Thus, the ratio of the actual current increase after the jump (R2 · t · I0) to the extrapolated current increase (R1 · t · I0) gives the ratio of channel formation rates (R2/R1).
Analysis of membrane potential of AZ-521 cells.
Experiments to analyze the membrane potential of AZ-521 cells were performed as described previously, with minor modifications (43, 47). Briefly, cells were detached with Accutase (Innovative Cell Technologies), washed, and then incubated with bis-(3-propyl-5-oxoisoxazol-4-yl)pentamethine oxonol (oxonol VI; Molecular Probes) at a final concentration of 2.5 μM for 30 min at 37°C. A cell suspension (2 ml) was placed in a stirred quartz cuvette at 37°C in a Perkin-Elmer Life Sciences LS50B fluorimeter. After stabilization of the fluorescence signal (excitation, 585 nm; emission, 645 nm), acid-activated VacA toxins (final concentration, 10 μg/ml) were added to the cells and changes in fluorescence were monitored. Depolarization causes a potential-dependent change in the cytoplasmic-transmembrane distribution of the fluorophore, which is accompanied by a change in fluorescence (2).
RESULTS
Identification of inactive VacA mutant proteins.
A nonspecific mutagenesis strategy was used to introduce mutations into the vacA gene. In this strategy, a plasmid (pMM592) expressing rVacA (31) was propagated in mutator E. coli strain XL1-Red (Stratagene). The XL1-Red strain is mutated in mutS, mutD, and mutT. The defects in these three DNA repair pathways result in an increased frequency of mutations, including transitions, transversions, insertions, and deletions (Stratagene). Colonies from the transformation into XL1-Red were pooled, and plasmid DNA was extracted. The resulting library of mutated plasmids was then transformed into E. coli expression strain ER2566, which encodes an IPTG-inducible copy of the RNA polymerase gene from bacteriophage T7. ER2566 transformants were cultured as described in Materials and Methods, in order to induce VacA expression. Soluble extracts from 4,249 individual colonies were prepared and analyzed. The mutation frequency in XL1-Red has been estimated at 1 in 2,000 bp/30 generations (21). At this mutation frequency, assuming that the mutations are randomly distributed, the Poisson distribution predicts that the probability of having no mutation in any 3-bp region of the vacA gene among the library of 4,249 mutant plasmids is <0.2%.
To examine the vacuolating activity of the mutant VacA proteins, an aliquot from the total cell lysate of each of the 4,249 samples was added to the medium overlying HeLa cells. Vacuolation of HeLa cells was assessed by microscopic inspection. Mutants that did not exhibit vacuolating activity based on microscopic observation were examined in more detail. All mutant proteins identified in the initial screen as lacking cytotoxic activity were immunoblotted with an anti-VacA antiserum to assess levels of VacA expression and the sizes of the mutant VacA proteins (31, 43). Mutants expressing VacA proteins that were substantially degraded, truncated, insoluble, or expressed at markedly reduced levels relative to a wild-type control were not studied further. Mutants that expressed VacA proteins that appeared to be full length and present in a quantity similar to that of the wild-type control were retested. New cultures of these mutants were grown, toxin concentrations were normalized based on an antibody capture ELISA, and equal amounts of wild-type or mutant rVacA proteins were added in triplicate to HeLa cell monolayers. Based on this analysis, 10 mutants were identified that failed to induce vacuolation (Fig. 1).
FIG. 1.

Analysis of mutant rVacA proteins that lack vacuolating activity. Plasmid pMM592, which expresses wild-type rVacA (31), was subjected to random mutagenesis by propagation in E. coli strain XL1-Red. Mutated plasmid DNA was isolated and introduced into expression strain ER2566. Soluble extracts from 4,249 individual colonies were screened for the ability to induce cytoplasmic vacuolation of HeLa cells. From this analysis, 10 mutants were isolated that failed to induce vacuolation. Results represent the vacuolating activity of soluble extracts from IPTG-induced ER2566 expressing wild-type rVacA, the 10 mutant proteins, and nonvacuolating rVacAΔ(6-27) (31, 55). Vacuolating activity was measured by neutral red uptake, and the mean and standard deviation of triplicate samples are shown (9). Results are expressed as a percentage of neutral red uptake relative to that of cells treated with wild-type rVacA.
The DNA sequence of the vacA gene was determined for each of the 10 mutants depicted in Fig. 1. Each mutant contained a single nucleotide polymorphism within the vacA gene, and each of these was a missense mutation (Fig. 2). Among the 10 mutant plasmids, eight different amino acid positions within VacA were mutated; 2 different mutations were identified at one position (S25P and S25L mutations at amino acid 25), and duplicate copies of the same mutation were identified at amino acid 18 (G18S). Of the 10 mutations identified, 6 map within the amino-terminal hydrophobic region of VacA, a region known to be important for toxin activity (12, 25, 26, 30, 33, 62, 63); 2 of the 6 mutations within the hydrophobic region are at amino acid positions that have previously been demonstrated to be important for VacA activity (P9 and G18) (33, 62). The remaining four mutations map outside of this hydrophobic region and have not previously been characterized. Curiously, all 10 mutations mapped within the p33 domain of VacA and no mutations were identified that mapped within the p55 domain of VacA (Fig. 2).
FIG. 2.

Mutations that inactivate rVacA. Plasmid DNA was isolated from each of the 10 mutants depicted in Fig. 1, and DNA sequence analysis was performed. Each mutant had a single missense mutation. For example, in isolate 1, the wild-type proline at amino acid position 9 was changed to serine (P9S); isolate 2, T152A; isolate 3, G18S; isolate 4, G18S; isolate 5, T210A; isolate 6, S25P; isolate 7, G121R, isolate 8, S246L; isolate 9, V21L; isolate 10, S25L. The relative positions of the 10 amino acid substitutions that resulted in loss of rVacA activity are shown. The amino-terminal p33 (amino acids 1 to 312 of the mature toxin, open rectangle) and carboxy-terminal p55 (amino acids 313 to 821 of the mature toxin, filled rectangle) domains of the 821-amino-acid mature VacA toxin are also shown.
Construction of H. pylori mutant strains.
To facilitate further analysis, six mutations (V21L, S25L, G121R, T152A, T210A, and S246L), each at an amino acid position not previously shown to be critical for VacA activity, were introduced into the H. pylori vacA gene by allelic exchange (4, 30, 33, 55). Each of the mutant strains secreted an 88-kDa VacA protein into the culture supernatant, based on detection by immunoblotting (data not shown). These mutant VacA proteins were then purified from the culture supernatants as described in Materials and Methods. Each of the mutant proteins eluted in fractions previously shown to contain high-molecular-mass oligomeric VacA from wild-type H. pylori, suggesting that none of the mutant proteins was defective in the formation of oligomeric complexes (data not shown). Purified VacA proteins were normalized based on an ELISA with anti-VacA serum, and equal amounts of VacA proteins were added to cell monolayers to test for vacuolating activity (Fig. 3). In agreement with the analysis of the mutant VacA proteins expressed in E. coli, VacA proteins carrying the V21L, S25L, G121R, or S246L mutation exhibited impaired vacuolating activity. In contrast, when expressed in H. pylori, VacA-T152A and VacA-T210A caused cell vacuolation similar to that produced by wild-type VacA (data not shown), a result that differed from the analysis of these two mutant proteins in the E. coli expression system. In summary, these experiments confirmed that four previously uncharacterized mutant VacA proteins containing the V21L, S25L, G121R, or S246L mutation each had impaired vacuolating activity.
FIG. 3.

Mutations that inactivate H. pylori VacA. Mutations at sites not previously shown to be critical for VacA activity were introduced into the H. pylori vacA gene by allelic exchange (4, 30, 33, 55). The VacA proteins were purified from the broth culture supernatants of wild-type (wt) and mutant strains and normalized based on an ELISA with anti-VacA serum (8, 54). Equal amounts of VacA proteins (▪, wild-type VacA; □, VacA-V21L; •, VacA-S25L; ○, VacA-G121R; ▴, VacA-S246L) were added to HeLa cell monolayers, and the cells were incubated for 16 h. Vacuolation was measured by neutral red uptake assay (9). Results represent the mean and standard deviation from triplicate samples. (Inset) Silver-stained gel of 88-kDa VacA proteins purified from H. pylori culture supernatant.
Binding and internalization of mutant VacA proteins.
Vacuolating cytotoxic activity is dependent on the ability of the toxin to bind to cells and be internalized by cells (6, 32, 36). Therefore, we tested each of the inactive mutant proteins for these properties. For these experiments, we analyzed purified mutant VacA proteins (V21L, S25L, G121R, and S246L) isolated from H. pylori culture supernatants. Each of the four mutant proteins bound to HeLa cells (Fig. 4), and a modest reduction in binding of two mutant proteins compared to wild-type VacA was detected. All of the mutant proteins were internalized by HeLa cells as determined by confocal microscopy (Fig. 5), which suggests that the modest reductions in binding observed for two of the mutant proteins were probably not biologically significant.
FIG. 4.

Binding of wild-type (wt) and mutant VacA to HeLa cells. VacA proteins (2.5 μg/ml) purified from H. pylori were incubated with HeLa cell monolayers at 4°C for 1 h. Monolayers were then washed to remove unbound toxin and fixed in 50% acetone-50% methanol. Bound VacA was then detected by ELISA with anti-VacA serum and horseradish peroxidase-labeled anti-rabbit immunoglobulin G. Net optical density at 450 nm was calculated by subtracting results obtained with control cells incubated in medium alone (no VacA). Results represent the mean and standard deviation of six or more samples. Comparisons between each mutant and the wild-type control were made by analysis of variance and Dunnett's post-hoc test. Asterisks denote results significantly different from those of the wild type (P < 0.05).
FIG. 5.
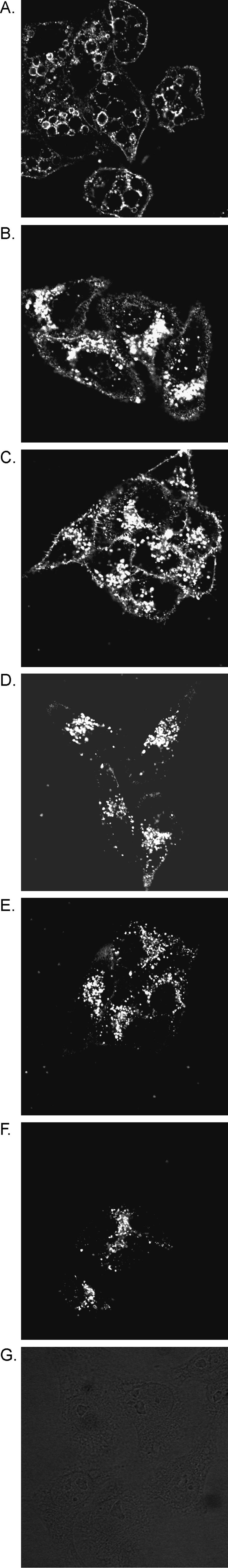
VacA proteins are internalized by HeLa cells. Equal amounts of VacA proteins (A, wild-type VacA; B, wild-type VacA in the absence of ammonium chloride; C, VacA-V21L; D, VacA-S25L; E, VacA-G121R; F, VacA-S246L; G, HeLa cells alone) were incubated with HeLa cells on glass coverslips at 37°C for 4 h. In each case, except panel B, the tissue culture medium was supplemented with 5 mM ammonium chloride. Cells were then fixed, permeabilized, and incubated with anti-VacA antiserum and Cy-3-labeled secondary antibody as described in Materials and Methods. The coverslips were then mounted on microscope slides, and the cells were visualized by confocal microscopy as described in Materials and Methods.
Protein dimerization.
We previously investigated functional properties of the amino-terminal hydrophobic portion of VacA with a TOXCAT model system and showed that the amino-terminal 32 amino acids of VacA are able to mediate protein dimerization (29, 33, 40, 41). In the TOXCAT model system, dimerization of a fusion protein results in expression of the cat gene, which is under the control of the dimerization-dependent transcription activator ToxR (reviewed in reference 14). We previously showed that the inability of certain mutant versions of this 32-amino-acid VacA peptide to mediate protein dimerization was correlated with the inability of the corresponding mutant VacA proteins to induce vacuolation and to form ion-conductive channels (33). We therefore examined the effect of the V21L and S25L mutations on protein dimerization. Neither mutation inhibited the capacity of the 32 amino-terminal amino acids of VacA to mediate protein dimerization in this system (Fig. 6). In contrast, a G14A mutation did interfere with protein dimerization, as expected based on previous studies (29, 33).
FIG. 6.

Protein dimerization mediated by the VacA N-terminal region. E. coli MM39/pccVacA-wt, MM39/pccVacA-G14A, MM39/pccVacA-V21L, and MM39/pccVacA-S25L were cultured in Luria-Bertani medium to an optical density 600 nm of approximately 0.35. Plasmids pccVacA-wt and pccVacA-G14A have been described previously (29), and the other two plasmids were constructed by similar methods. CAT activity from each strain was quantified by CAT ELISA (Roche). Results represent the mean and standard deviation from triplicate cultures. Comparisons between each mutant and the wild-type (wt) control were made by analysis of variance and Dunnett's post-hoc test. Asterisks denote results significantly different from those of the wild type (P < 0.05).
Membrane channel formation by mutant VacA proteins.
Vacuolating cytotoxic activity is likely to be dependent on the ability of VacA to form membrane channels in cellular membranes (10, 23, 33, 47, 57). To characterize the channel-forming properties of the mutant VacA proteins, purified proteins were analyzed by planar lipid bilayer assays. The two mutant proteins containing the V21L and S25L mutations, which map within the amino-terminal hydrophobic region, did not form stable channels (Table 1). This is consistent with the failure of several other inactive VacA mutant proteins containing mutations within the amino-terminal hydrophobic region to form stable channels (30, 33, 55). The other two mutant VacA proteins, carrying the G121R or S246L mutation, formed channels similar to those formed by wild-type VacA. These are the first examples of mutant VacA proteins that exhibit channel activities similar to that of wild-type VacA but fail to induce vacuole formation in eukaryotic cells. Additional studies indicated that VacA-G121R and VacA-S246L formed channels at a slightly faster rate than wild-type VacA at a positive membrane potential (Table 1).
Membrane depolarization.
VacA is known to form anion-selective channels in the plasma membrane of cells, and the formation of these channels results in partial depolarization of the resting membrane potential (33, 43, 47). To determine whether the VacA-V21L, -S25L, -G121R, and -S246L mutant proteins were defective in the ability to cause membrane depolarization, we used bis-(3-propyl-5-oxoisoxazol-4-yl)pentamethine oxonol as a probe to monitor the membrane potential of AZ521 cells. Consistent with previously published results (33, 43, 47), we found that the addition of acid-activated wild-type VacA rapidly altered the resting membrane potential of AZ521 cells, whereas the resting membrane potential was not altered by acid-activated VacA-G18A (Fig. 7) (33). Similarly, the resting membrane potential of AZ521 cells was not altered by either VacA-V21L or VacA-S25L. Addition of VacA-G121R or VacA-S246L to cells resulted in depolarization of the resting membrane potential, consistent with the bilayer assays demonstrating the channel-forming capacity of these mutants. Similar results were obtained with HeLa cells (data not shown). These are the first examples of mutant VacA proteins that retain the capacity to cause membrane depolarization yet fail to cause cell vacuolation.
FIG. 7.

Analysis of membrane depolarization induced by mutant VacA proteins. VacA proteins were purified from H. pylori broth culture supernatants as described in Materials and Methods. AZ521 cells were loaded with oxonol VI (a probe used to monitor membrane potential). Following addition of acid-activated VacA proteins (10 μg/ml), changes in fluorescence were monitored. The arrow indicates the time at which toxin was added to the cuvette. An increase in fluorescence (relative fluorescence units [RFU]) over time indicates depolarization of the membrane potential. wt, wild type.
DISCUSSION
In this study, we identified 10 mutant VacA proteins that lacked vacuolating toxin activity when added to HeLa cells. All of the mutations identified in the present study mapped within the p33 domain of VacA. Previous studies have shown that amino acid sequences located within a hydrophobic region near the amino terminus of the p33 domain are required for the formation of anion-selective membrane channels (30, 33), and regions within the p55 domain are required for VacA binding to mammalian cells (17, 39, 56). Potentially, the failure to identify inactivating mutations within the p55 domain is attributable to the ability of VacA to bind multiple cell surface receptors (10, 20, 34, 35, 44, 53, 58-60). Alternatively, binding of VacA to mammalian cells may be mediated by a region of the VacA protein comprising multiple amino acids and mutagenesis of individual residues may be insufficient to ablate the binding properties of VacA.
Two of the 10 mutations identified in the present study (T152A and T210A) resulted in loss of vacuolating activity when the mutant proteins were expressed in E. coli but did not disrupt VacA activity when these mutant proteins were expressed in H. pylori. It is possible that factors present in H. pylori but absent from E. coli promote the proper folding of these mutant VacA proteins. For example, proper VacA folding may be promoted by H. pylori chaperone proteins or by the carboxy-terminal autotransporter domain of VacA that is present in H. pylori but absent from the rVacA proteins expressed in this study (16). Alternatively, it is possible that these two mutant proteins, when expressed in E. coli, underwent degradation to yield dominant-negative mutant forms of VacA, whereas such degradation did not occur when the proteins were expressed in H. pylori (55).
The fact that 6 of 10 mutations identified in the screen of mutant rVacA proteins map within the amino-terminal hydrophobic region (Fig. 2) underscores the importance of this region in VacA activity and indicates that the activity of this region can be readily ablated following mutation of individual amino acid residues (33, 62). As shown in this study, the V21L and S25L VacA mutant proteins exhibit characteristics similar to those of previously characterized P9A, G14A, and G18A mutant proteins (33, 62). These similarities include defects in the ability to induce cytoplasmic vacuolation, defects in the ability to induce depolarization of the plasma membrane, and defects in the ability to form anion-conductive channels in lipid bilayers. It has been hypothesized that the amino-terminal hydrophobic region of VacA directly inserts into membranes to form the VacA channel (24, 33). A theoretical structural model of the amino-terminal end of VacA proposes that this region consists of a hexameric helical bundle (24). In this model, amino acids such as G14 and G18 (which are critical for toxin activity and for the ability of this region of VacA to dimerize in the TOXCAT model of transmembrane protein interaction) (29, 33, 40) are positioned at the interface between adjacent helices. Conversely, this model proposes that amino acids such as P9, V21, and S25 (which are not required for protein dimerization in the TOXCAT model) are not positioned at the interface between adjacent helices.
The G121R and S246L mutations identified in this study represent the first single amino acid substitutions that reduce vacuolating cytotoxic activity and that map outside the amino-terminal hydrophobic region. In an effort to determine why these two mutant toxins were inactive, we analyzed five properties that are thought to be required for vacuolating toxin activity. The G121R and S246L mutant toxins were secreted by H. pylori and assembled into water-soluble oligomeric structures similar in size to those formed by wild-type VacA. The VacA-G121R and VacA-S246L mutant toxins bound to cells, were internalized by cells, and caused depolarization of intact cells. Both mutant proteins formed channels in planar lipid bilayers that exhibited an anion selectivity similar to that of channels formed by wild-type VacA. Thus, these two mutant toxins were not defective in any of the activities currently known to be required for vacuolating toxin activity. The retention of these functional properties suggests that these mutant toxins were not misfolded. Retention of membrane channel-forming activity by VacA-G121R and -S246L distinguishes these mutant proteins from those with mutations that map within the amino-terminal hydrophobic region. The defective vacuolating activity of VacA-G121R and -S246L mutant proteins, despite retention of membrane channel-forming activity, supports our conclusion from a previous study that channel formation is necessary but not sufficient for vacuolation (30).
Although the VacA-G121R and -S246L proteins formed channels at a rate similar to that of wild-type VacA at a negative membrane potential, these mutant proteins formed channels at a slightly faster rate than wild-type VacA at a positive membrane potential (Table 1). The plasma membrane of a cell typically has a transmembrane potential of approximately −70 mV (negative inside) as a consequence of K+, Na+, and Cl− concentration gradients that are maintained by active transport processes (22). Based on this positive-outside membrane potential and assuming that VacA has a similar affinity for cells and lipid bilayers, it might be expected that VacA-G121R and -S246L mutant proteins would induce a more rapid depolarization of cells than wild-type VacA, yet this was not observed. It should be noted that the conditions for the planar lipid bilayer assays and the cell depolarization assays were not identical. Specifically, the bilayer studies were performed at a lower pH than the VacA channel likely experiences when it causes depolarization of cells. Moreover, the kinetics of cell depolarization were quite different from the rise in current observed in the bilayer assay, which suggests that there are substantial differences in the VacA-mediated events that are measured in the two assays.
The exact functional role of VacA residues G121 and S246 in vacuolating toxin activity remains unclear. It is possible that the relatively subtle differences in the channel activities of wild-type VacA compared to the VacA-G121R and -S246L mutant proteins might account for the failure of these mutant toxins to cause cell vacuolation. Alternatively, we speculate that the VacA-G121R and -S246L mutant toxins might be defective in the ability to interact with specific cellular target molecules or might be defective in one or more uncharacterized intracellular events required for cell vacuolation.
Acknowledgments
This work was supported by the National Institutes of Health (R01 AI39657 and R01 DK53623) and the Department of Veterans Affairs.
We thank Valerie Busler and Beverly Hosse for technical assistance.
Editor: J. T. Barbieri
Footnotes
Published ahead of print on 5 September 2006.
REFERENCES
- 1.Al-Awqati, Q. 1986. Proton-translocating ATPases. Annu. Rev. Cell Biol. 2:179-199. [DOI] [PubMed] [Google Scholar]
- 2.Apell, H. J., and B. Bersch. 1987. Oxonol VI as an optical indicator for membrane potentials in lipid vesicles. Biochim. Biophys. Acta 903:480-494. [DOI] [PubMed] [Google Scholar]
- 3.Boncristiano, M., S. R. Paccani, S. Barone, C. Ulivieri, L. Patrussi, D. Ilver, A. Amedei, M. M. D'Elios, J. L. Telford, and C. T. Baldari. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198:1887-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Copass, M., G. Grandi, and R. Rappuoli. 1997. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect. Immun. 65:1949-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cover, T. L., D. E. Berg, M. J. Blaser, and H. L. T. Mobley. 2001. H. pylori pathogenesis, p. 510-558. In E. A. Groisman (ed.), Principles of bacterial pathogenesis. Academic Press, San Diego, Calif.
- 6.Cover, T. L., and S. R. Blanke. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320-332. [DOI] [PubMed] [Google Scholar]
- 7.Cover, T. L., and M. J. Blaser. 1992. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 267:10570-10575. [PubMed] [Google Scholar]
- 8.Cover, T. L., P. I. Hanson, and J. E. Heuser. 1997. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 138:759-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cover, T. L., W. Puryear, G. I. Pérez-Pérez, and M. J. Blaser. 1991. Effect of urease on HeLa cell vacuolation induced by Helicobacter pylori cytotoxin. Infect. Immun. 59:1264-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czajkowsky, D. M., H. Iwamoto, T. L. Cover, and Z. Shao. 1999. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl. Acad. Sci. USA 96:2001-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czajkowsky, D. M., H. Iwamoto, G. Szabo, T. L. Cover, and Z. Shao. 2005. Mimicry of a host anion channel by a Helicobacter pylori pore-forming toxin. Biophys. J. 89:3093-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Bernard, M., D. Burroni, E. Papini, R. Rappuoli, J. Telford, and C. Montecucco. 1998. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect. Immun. 66:6014-6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Bernard, M., E. Papini, V. de Filippis, E. Gottardi, J. Telford, R. Manetti, A. Fontana, R. Rappuoli, and C. Montecucco. 1995. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 270:23937-23940. [DOI] [PubMed] [Google Scholar]
- 14.DiRita, V. J. 1992. Co-ordinate expression of virulence genes by ToxR in Vibrio cholerae. Mol. Microbiol. 6:451-458. [DOI] [PubMed] [Google Scholar]
- 15.Dunn, B. E., H. Cohen, and M. J. Blaser. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer, W., R. Buhrdorf, E. Gerland, and R. Haas. 2001. Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect. Immun. 69:6769-6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garner, J. A., and T. L. Cover. 1996. Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect. Immun. 64:4197-4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gauthier, N. C., V. Ricci, L. Landraud, and P. Boquet. 2006. Helicobacter pylori VacA toxin: a tool to study novel early endosomes. Trends Microbiol. 14:292-294. [DOI] [PubMed] [Google Scholar]
- 19.Gebert, B., W. Fischer, E. Weiss, R. Hoffmann, and R. Haas. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099-1102. [DOI] [PubMed] [Google Scholar]
- 20.Geisse, N. A., T. L. Cover, R. M. Henderson, and J. M. Edwardson. 2004. Targeting of Helicobacter pylori vacuolating toxin to lipid raft membrane domains analysed by atomic force microscopy. Biochem. J. 381:911-917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greener, A., M. Callahan, and B. Jerpseth. 1996. An efficient random mutagenesis technique using an E. coli mutator strain. Methods Mol. Biol. 57:375-385. [DOI] [PubMed] [Google Scholar]
- 22.Hille, B. 2001. Ion channels of excitable membranes, third ed. Sinauer Associates, Inc., Sunderland, Mass.
- 23.Iwamoto, H., D. M. Czajkowsky, T. L. Cover, G. Szabo, and Z. Shao. 1999. VacA from Helicobacter pylori: a hexameric chloride channel. FEBS Lett. 450:101-104. [DOI] [PubMed] [Google Scholar]
- 24.Kim, S., A. K. Chamberlain, and J. U. Bowie. 2004. Membrane channel structure of Helicobacter pylori vacuolating toxin: role of multiple GXXXG motifs in cylindrical channels. Proc. Natl. Acad. Sci. USA 101:5988-5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Letley, D. P., and J. C. Atherton. 2000. Natural diversity in the N terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J. Bacteriol. 182:3278-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Letley, D. P., J. L. Rhead, R. J. Twells, B. Dove, and J. C. Atherton. 2003. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J. Biol. Chem. 278:26734-26741. [DOI] [PubMed] [Google Scholar]
- 27.Leunk, R. D., P. T. Johnson, B. C. David, W. G. Kraft, and D. R. Morgan. 1988. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 26:93-99. [DOI] [PubMed] [Google Scholar]
- 28.Li, X., T. Wang, Z. Zhao, and S. A. Weinman. 2002. The ClC-3 chloride channel promotes acidification of lysosomes in CHO-K1 and Huh-7 cells. Am. J. Physiol. Cell Physiol. 282:C1483-C1491. [DOI] [PubMed] [Google Scholar]
- 29.McClain, M. S., P. Cao, and T. L. Cover. 2001. Amino-terminal hydrophobic region of Helicobacter pylori vacuolating cytotoxin (VacA) mediates transmembrane protein dimerization. Infect. Immun. 69:1181-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McClain, M. S., P. Cao, H. Iwamoto, A. D. Vinion-Dubiel, G. Szabo, Z. Shao, and T. L. Cover. 2001. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J. Bacteriol. 183:6499-6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClain, M. S., and T. L. Cover. 2003. Expression of Helicobacter pylori vacuolating toxin in Escherichia coli. Infect. Immun. 71:2266-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McClain, M. S., and T. L. Cover. 2006. Helicobacter pylori vacuolating toxin, p. 468-490. In J. E. Alouf and M. R. Popoff (ed.), The comprehensive sourcebook of bacterial protein toxins, 3rd ed. Elsevier, Amsterdam, The Netherlands.
- 33.McClain, M. S., H. Iwamoto, P. Cao, A. D. Vinion-Dubiel, Y. Li, G. Szabo, Z. Shao, and T. L. Cover. 2003. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 278:12101-12108. [DOI] [PubMed] [Google Scholar]
- 34.Molinari, M., C. Galli, M. de Bernard, N. Norais, J. M. Ruysschaert, R. Rappuoli, and C. Montecucco. 1998. The acid activation of Helicobacter pylori toxin VacA: structural and membrane binding studies. Biochem. Biophys. Res. Commun. 248:334-340. [DOI] [PubMed] [Google Scholar]
- 35.Moll, G., E. Papini, R. Colonna, D. Burroni, J. Telford, R. Rappuoli, and C. Montecucco. 1995. Lipid interaction of the 37-kDa and 58-kDa fragments of the Helicobacter pylori cytotoxin. Eur. J. Biochem. 234:947-952. [DOI] [PubMed] [Google Scholar]
- 36.Montecucco, C., and R. Rappuoli. 2001. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell. Biol. 2:457-466. [DOI] [PubMed] [Google Scholar]
- 37.Morbiato, L., F. Tombola, S. Campello, G. Del Giudice, R. Rappuoli, M. Zoratti, and E. Papini. 2001. Vacuolation induced by VacA toxin of Helicobacter pylori requires the intracellular accumulation of membrane permeant bases, Cl− and water. FEBS Lett. 508:479-483. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen, V. Q., R. M. Caprioli, and T. L. Cover. 2001. Carboxy-terminal proteolytic processing of Helicobacter pylori vacuolating toxin. Infect. Immun. 69:543-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reyrat, J. M., S. Lanzavecchia, P. Lupetti, M. de Bernard, C. Pagliaccia, V. Pelicic, M. Charrel, C. Ulivieri, N. Norais, X. Ji, V. Cabiaux, E. Papini, R. Rappuoli, and J. L. Telford. 1999. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 290:459-470. [DOI] [PubMed] [Google Scholar]
- 40.Russ, W. P., and D. M. Engelman. 2000. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 296:911-919. [DOI] [PubMed] [Google Scholar]
- 41.Russ, W. P., and D. M. Engelman. 1999. TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. USA 96:863-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salama, N. R., G. Otto, L. Tompkins, and S. Falkow. 2001. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun. 69:730-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schraw, W., Y. Li, M. S. McClain, F. G. van der Goot, and T. L. Cover. 2002. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 277:34642-34650. [DOI] [PubMed] [Google Scholar]
- 44.Seto, K., Y. Hayashi-Kuwabara, T. Yoneta, H. Suda, and H. Tamaki. 1998. Vacuolation induced by cytotoxin from Helicobacter pylori is mediated by the EGF receptor in HeLa cells. FEBS Lett. 431:347-350. [DOI] [PubMed] [Google Scholar]
- 45.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175-1186. [DOI] [PubMed] [Google Scholar]
- 46.Sundrud, M. S., V. J. Torres, D. Unutmaz, and T. L. Cover. 2004. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 101:7727-7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szabo, I., S. Brutsche, F. Tombola, M. Moschioni, B. Satin, J. L. Telford, R. Rappuoli, C. Montecucco, E. Papini, and M. Zoratti. 1999. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18:5517-5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Telford, J. L., P. Ghiara, M. Dell'Orco, M. Comanducci, D. Burroni, M. Bugnoli, M. F. Tecce, S. Censini, A. Covacci, Z. Xiang, E. Papini, C. Montecucco, L. Parente, and R. Rappuoli. 1994. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179:1653-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tombola, F., C. Carlesso, I. Szabo, M. de Bernard, J. M. Reyrat, J. L. Telford, R. Rappuoli, C. Montecucco, E. Papini, and M. Zoratti. 1999. Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: possible implications for the mechanism of cellular vacuolation. Biophys. J. 76:1401-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torres, V. J., S. E. Ivie, M. S. McClain, and T. L. Cover. 2005. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 280:21107-21114. [DOI] [PubMed] [Google Scholar]
- 51.Torres, V. J., M. S. McClain, and T. L. Cover. 2004. Interactions between p-33 and p-55 domains of the Helicobacter pylori vacuolating cytotoxin (VacA). J. Biol. Chem. 279:2324-2331. [DOI] [PubMed] [Google Scholar]
- 52.Torres, V. J., M. S. McClain, and T. L. Cover. 2006. Mapping of a domain required for protein-protein interactions and inhibitory activity of a Helicobacter pylori dominant-negative VacA mutant protein. Infect. Immun. 74:2093-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Utt, M., B. Danielsson, and T. Wadstrom. 2001. Helicobacter pylori vacuolating cytotoxin binding to a putative cell surface receptor, heparan sulfate, studied by surface plasmon resonance. FEMS Immunol. Med. Microbiol. 30:109-113. [DOI] [PubMed] [Google Scholar]
- 54.Vinion-Dubiel, A. D., M. S. McClain, P. Cao, R. L. Mernaugh, and T. L. Cover. 2001. Antigenic diversity among Helicobacter pylori vacuolating toxins. Infect. Immun. 69:4329-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinion-Dubiel, A. D., M. S. McClain, D. M. Czajkowsky, H. Iwamoto, D. Ye, P. Cao, W. Schraw, G. Szabo, S. R. Blanke, Z. Shao, and T. L. Cover. 1999. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 274:37736-37742. [DOI] [PubMed] [Google Scholar]
- 56.Wang, H. J., and W. C. Wang. 2000. Expression and binding analysis of GST-vacA fusions reveals that the C-terminal approximately 100-residue segment of exotoxin is crucial for binding in HeLa cells. Biochem. Biophys. Res. Commun. 278:449-454. [DOI] [PubMed] [Google Scholar]
- 57.Willhite, D. C., T. L. Cover, and S. R. Blanke. 2003. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J. Biol. Chem. 278:48204-48209. [DOI] [PubMed] [Google Scholar]
- 58.Yahiro, K., T. Niidome, T. Hatakeyama, H. Aoyagi, H. Kurazono, P. I. Padilla, A. Wada, and T. Hirayama. 1997. Helicobacter pylori vacuolating cytotoxin binds to the 140-kDa protein in human gastric cancer cell lines, AZ-521 and AGS. Biochem. Biophys. Res. Commun. 238:629-632. [DOI] [PubMed] [Google Scholar]
- 59.Yahiro, K., T. Niidome, M. Kimura, T. Hatakeyama, H. Aoyagi, H. Kurazono, K. Imagawa, A. Wada, J. Moss, and T. Hirayama. 1999. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase beta. J. Biol. Chem. 274:36693-36699. [DOI] [PubMed] [Google Scholar]
- 60.Yahiro, K., A. Wada, M. Nakayama, T. Kimura, K. Ogushi, T. Niidome, H. Aoyagi, K. Yoshino, K. Yonezawa, J. Moss, and T. Hirayama. 2003. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 278:19183-19189. [DOI] [PubMed] [Google Scholar]
- 61.Ye, D., and S. R. Blanke. 2002. Functional complementation reveals the importance of intermolecular monomer interactions for Helicobacter pylori VacA vacuolating activity. Mol. Microbiol. 43:1243-1253. [DOI] [PubMed] [Google Scholar]
- 62.Ye, D., and S. R. Blanke. 2000. Mutational analysis of the Helicobacter pylori vacuolating toxin amino terminus: identification of amino acids essential for cellular vacuolation. Infect. Immun. 68:4354-4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye, D., D. C. Willhite, and S. R. Blanke. 1999. Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J. Biol. Chem. 274:9277-9282. [DOI] [PubMed] [Google Scholar]