

Abstract
Although the physiological function of the prion protein remains unknown, in vitro experiments suggest that the protein may bind copper (II) ions and play a role in copper transport or homoeostasis in vivo. The unstructured N-terminal region of the prion protein has been shown to bind up to six copper (II) ions, with each of these ions co-ordinated by a single histidine imidazole and nearby backbone amide nitrogen atoms. Individually, these sites have micromolar affinities, which is weaker than would be expected of a true cuproprotein. In the present study, we show that with subsaturating levels of copper, different forms of co-ordination will occur, which have higher affinity. We have investigated the copper-binding properties of two peptides representing the known copper-binding regions of the prion protein: residues 57–91, which contains four tandem repeats of the octapeptide GGGWGQPH, and residues 91–115. Using equilibrium dialysis and spectroscopic methods, we unambiguously demonstrate that the mode of copper co-ordination in both of these peptides depends on the number of copper ions bound and that, at low copper occupancy, copper ions are co-ordinated with sub-micromolar affinity by multiple histidine imidazole groups. At pH 7.4, three different modes of copper co-ordination are accessible within the octapeptide repeats and two within the peptide comprising residues 91–115. The highest affinity copper (II)-binding modes cause self-association of both peptides, suggesting a role for copper (II) in controlling prion protein self-association in vivo.
Keywords: copper (II), equilibrium dialysis, prion protein (PrP), self-association
Abbreviations: CSF, cerebrospinal fluid; ICP, inductively coupled plasma; NEM, N-ethylmorpholine; PrP, prion protein; PrPC, PrP cellular isoform; PrP57–91 and PrP91–115, peptides representing residues 57–91 and residues 91–115 of the human prion protein respectively; Cu2(PrP57–91), complex of PrP57–91 with two copper ions; Cu(PrP91–115)2, complex of a copper ion with two PrP91–115 molecules
INTRODUCTION
Prion diseases are a group of transmissible neurodegenerative disorders, found in both humans and animals, which have become the focus of intense scientific interest because of their apparently unique underlying biology [1]. The ‘protein-only’ hypothesis is now widely accepted and proposes that the causative agent of these diseases is largely, if not exclusively, composed of an abnormal conformer of a host-encoded protein known as the prion protein, PrP, and is devoid of nucleic acid [2,3]. Mature human PrP is a 209-residue polypeptide with a C-terminal glycosylphosphatidylinositol anchor and two glycosylation sites, that comprises a folded C-terminal domain and a flexibly disordered N-terminal region [4–6]. Although much is now known about the disease-causing properties of the prion protein, the physiological function of the protein remains an enigma. PrP is expressed in most tissues in adults, but the highest levels of expression occur in the central nervous system and the immune system [7,8]. Transgenic mice in which the Prnp gene has been disrupted and hence PrPC (PrP cellular isoform) expression is ablated display no developmental or behavioural phenotype [9], although more detailed studies have described electrophysiological abnormalities [10] and alterations in sleep and circadian rhythms [11].
In vitro experiments have revealed that the prion protein specifically binds copper (II) [12–15] and this had led to the proposal that PrP may function as a cuproprotein in vivo. However, the evidence for this assertion is limited; mice that do not express PrP have been reported to have reduced levels of copper (II) in membrane fractions from the brain [16], although this result could not be replicated by other investigators [17]. Cultured cells induced to express PrP have been shown to have a higher copper-binding capacity than uninduced cells [18]. Copper (II) has also been reported to cause internalization of PrP, but the concentrations required to cause this effect are vastly above measured physiological levels [18,19].
A clear understanding of the copper (II)-binding properties of the prion protein in vitro is thus essential in order to critically evaluate proposals relating to any copper-binding role in vivo. The interaction of full-length and truncated forms of the prion protein with copper (II) has been investigated using a range of techniques including EPR [20–22], CD [23], X-ray crystallography [24], NMR [25,26], MS [14,15], Raman spectroscopy [27,28], FTIR (Fourier transform IR) spectroscopy [29] and potentiometry [30]. A consensus has emerged that, at maximum copper (II) occupancy, the prion protein can bind five or six copper ions, and that each of these ions is co-ordinated by a single histidine imidazole and two deprotonated backbone amide nitrogen atoms [14,21,24]. In this way, each of the four octapeptide repeats can bind a single ion, and sites at His96 and His111 can bind an additional two ions in a similar fashion [21,26]. It has, however, become clear that, at lower copper (II) occupancy, copper co-ordination by the octapeptide repeats is different to the total occupancy state. Three published works now report that, within the four octapeptide repeats, a single copper (II) ion can be co-ordinated by multiple histidine imidazoles without the involvement of backbone amides [25,30,31]. Most estimates of the affinity of PrP for copper have been in the low-micromolar range [15,23,32,33], but the affinity has also been placed dramatically higher, in the femtomolar range under idealized conditions [25]. In the absence of definitive in vivo experiments which might confirm that PrP is a true cuproprotein, in vitro affinity measurements offer an alternative route to assess whether this hypothesis is credible. In vivo, most extracellular copper is sequestered in complexes with proteins and amino acids which have affinities in the nanomolar-to-picomolar range [34,35], suggesting that, if PrP does have only a micromolar affinity for copper, it would be unlikely to bind copper under normal physiological conditions.
In the present study, we used equilibrium dialysis to thoroughly characterize the multiple modes of copper co-ordination which can occur in peptides representing residues 57–91 (PrP57–91, containing the four octapeptide repeats) and residues 91–115 (PrP91–115, containing His96 and His111) of the human prion protein. As well as allowing the determination of stoichiometry, this technique can be used to identify sites of different affinity through Scatchard analysis and provides a framework for the unambiguous interpretation of spectroscopic measurements. In order to keep copper (II) soluble above about pH 5, a chelating buffer must be used. As these buffers also compete against any interaction of copper (II) with the peptide, the experiment reports an apparent affinity that is weakened relative to the true value. In each case, the true dissociation constants has been calculated by determining the free copper concentration when the total copper concentration is the same as the apparent dissociation constant. We have used this information, in conjunction with CD, fluorescence and NMR spectroscopic measurements to characterize the multiple modes in which copper (II) can be co-ordinated by the PrP peptides. The data reveal that the affinities of the lower-occupancy copper-binding modes are in the nanomolar range. The present work demonstrates for the first time that multiple modes of copper (II) co-ordination can occur at the binding sites outside of the octapeptide repeats, and that self-association plays a role in the highest-affinity copper-binding mode of both peptides.
MATERIALS AND METHODS
Peptide synthesis and purification
Peptide synthesis was carried by ABC (Advanced Biotechnology Centre), Imperial College London, London, U.K. Peptides were produced by solid-phase stepwise synthesis using the Fmoc (fluoren-9-ylmethoxycarbonyl) N-terminal protection strategy. All peptide products were purified and analysed by reverse-phase HPLC. Molecular-mass determination was performed using MALDI (matrix-assisted laser-desorption ionization)-MS. Both peptides were acetylated at the N-terminus and amidated at the C-terminus. The peptide sequences used were PrP57–91, WGQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQ, and PrP91–115, QGGGTHSQWNKPSKPKTNMKHMAGA.
Equilibrium dialysis
Solutions of peptides in 5 mM Tris/HCl, pH 7.4, 50 mM Mops, pH 7.4, or 10 mM acetate buffer, pH 5.5, were dialysed for approx. 10 h against a 1 litre reservoir of copper-containing buffer, using Spectra/Por 7 3500 MWCO (molecular-mass cut-off) dialysis membrane. Copper was added to each buffer reservoir from a stock of either 20 mM CuSO4 or 20 mM CuSO4 containing 40 mM glycine, to achieve the desired copper concentration. After dialysis, samples of the peptide solution and the dialysis buffer were diluted 5-fold with 1% Aristar nitric acid, and the copper concentration then measured using a Spectro-Ciros CCD (charge-coupled device) ICP (inductively coupled plasma)-atomic emission spectrometer. Typically, final peptide concentrations were approx. 10 μM for PrP57–91 and approx. 40 μM for PrP91–115.
Measurements of peptide concentrations
Peptide concentrations were determined by measuring the absorbance of solutions at 280 nm using the molar absorption coefficient of tryptophan at that wavelength. For experiments on PrP91–115 at pH 7.4, the contribution of peptide-bound copper to the molar absorption coefficient at 280 nm was significant. The molar absorption coefficient of copper (II) in the Cu2(PrP91–115) complex (a complex of two copper ions with PrP91–115) was measured as 1700 M–1·cm–1, and a similar value was obtained for the Cu(PrP91–115)2 complex (a complex of a copper ion with two PrP91–115 molecules). The concentration of peptide bound copper was determined directly from the ICP-atomic emission spectrometry results, and thus the contribution of the peptide-bound copper to the absorbance at 280 nm could be calculated and subtracted from the raw data.
CD
Spectra were recorded on a Jasco J-810 spectropolarimeter at 25 °C, with a scanning speed of 10 nm/min and the slit width set to 150 μm. The signal-to-noise ratio was limited in most samples and so, to average out noise as much as possible, the data pitch was set to 5 nm, the integration time to 30 s per point and the number of scans per spectrum to four. Aliquots of 2 or 20 mM CuSO4 were added to peptide solutions in Tris and NEM (N-ethylmorpholine) buffers. For peptide samples in Mops buffer, copper was added as a glycine chelate either as 2 mM CuSO4 and 4 mM glycine, or 20 mM CuSO4 and 40 mM glycine. CD measurements in mdeg (θ) were converted into molar CD (Δϵ, with units of litre·mol–1·cm–1) using the relationship Δϵ=θ/(33000·l·c), where c is the concentration in mol/l and l is the pathlength in cm.
Fluorescence spectroscopy
Experiments were carried out on a Cary Eclipse fluorimeter at 25 °C, with the excitation and emission wavelengths set to 280 nm and 356 nm respectively. The excitation slit width was 5 μm for all experiments. The emission slit width was 10 μm for experiments on PrP57–91 and 5 μm for PrP91–115. The detector voltage was set to either 600 V or 800 V to bring the initial fluorescence of the sample into a measurable range. Peptide solutions were titrated with either 1 mM CuSO4 in 5 mM Tris/HCl, pH 7.4 (PrP57–91) or 10 mM CuSO4 in 50 mM Tris/HCl, pH 7.4 (for PrP91–115). Data were corrected for the dilution of the sample.
NMR
PrP57–91 samples contained 30 μM peptide in 25 mM phosphate buffer, pH 7.4, and were titrated with aliquots of 200 μM copper bisglycinate or 1 M Mops, pH 7.4. To exclude pH changes as a cause of the observed chemical-shift changes, the titration with Mops was repeated in 50 mM phosphate buffer, pH 7.4. The PrP91–115 sample contained 100 μM peptide in 5 mM Tris/HCl, pH 7.4, and was titrated with 1 mM CuSO4 in the same buffer. Both samples contained 5% 2H2O. Spectra of PrP57–91 were recorded on a Bruker DRX-600 spectrometer, whereas spectra of PrP91–115 were recorded on a DRX-500 instrument. Both spectrometers were equipped with cryoprobes. Water suppression was achieved by pre-saturation of the water resonance. The recycle delay was set to allow complete relaxation of the peptide resonances between scans. For PrP57–91, a 4 s recycle delay was used, whereas a 6 s delay was necessary for PrP91–115. All spectra were recorded at 25 °C.
Simulations of equilibria for formation of metal complexes and calculations of dissociation constants
Simulations were carried out using an in-house program to numerically calculate the equilibrium position of a series of linked equilibria, given fixed total concentrations of metal and ligand. In the simulations of binding to PrP57–91 shown in Figure 6, the four independent sites (termed mode C) were modelled using macroscopic dissociation constants of 0.25, 0.66, 1.5 and 4.0 μM, likewise the two independent sites (termed mode B) were modelled using macroscopic dissociation constants of 125 and 500 nM.
Figure 6. Simulation of three mutually exclusive copper (II)-binding modes in PrP57–91.

The concentrations of the main species present at equilibrium in simulations of competing modes of copper binding in PrP57–91 are shown over a range of total copper (II) concentrations. The simulations are described in detail in the text. They show that a single site of 3 nM affinity (mode A: CuAP) will predominate over four sites of 1 μM affinity (mode C: CuCP, CuC2P, etc.) only at low copper (II) concentrations. The concentrations of the CuBP and CuCP species did not exceed 1 μM at any point and are excluded for clarity. For illustration, an approximate predicted CD signal at 580 nM is shown, assuming that each copper (II) ion bound in modes B and C produces an equal signal at this wavelength and that ions bound in mode A produce no signal.
To obtain the non-linear fits to the Scatchard plots shown in Figures 7(B) and 7(C), the Kd values were first crudely estimated by iteratively adjusting the Kd parameters in the model. The final fit was obtained by running a suite of simulations to sample, to one significant figure, all of the possible combinations of the two or three Kd parameters. For example, for the data in Figure 7(B), the parameters were set to: 10, 20, 30, 40 and 50 μM for Kd(1); 3, 4, 5, 6 and 7 μM for Kd(2); and 20, 30, 40 and 50 μM for Kd(3), giving 100 data sets in total. The best fit to the observed data from these calculated data sets was then selected by least-squares fitting. The linear fits to the other Scatchard plots were also calculated by least-squares fitting.
Figure 7. Equilibrium dialysis reveals two distinct forms of copper (II) co-ordination in PrP91–115.

Scatchard plots of equilibrium dialysis data for PrP91–115 show copper (II) binding with stoichiometries of 0.5:1 or 2:1, depending on the solution conditions. (A) In 50 mM Mops buffer at pH 7.4, with copper presented as a glycine complex, two sites (intercept=2.11) of equal affinity are observed, each with an apparent dissociation constant of 5.5±0.5 μM. (B and C)The formation of a 0.5:1 complex gives a non-linear Scatchard plot. The data were fitted numerically as described in the text, the results of which are represented as continuous lines. Dialysis against CuSO4 in 5 mM Tris, pH 7.4 (B), showed formation of a Cu(PrP91–115)2 complex at low copper concentrations and a Cu2PrP91–115 complex at higher copper concentrations. In 10 mM acetate, pH 5.5, only the Cu(PrP91–115)2 complex was observed (C). (D) CD spectra could be observed for both the Cu(PrP91–115)2 and the Cu2(PrP91–115) complex. The spectra of the Cu(PrP91–115)2 complex formed in 5 mM Tris buffer, pH 7.4 (100 μM PrP91–115, 60 μM CuSO4, i.e. 0.5:1 CuSO4 and 10 μM free CuSO4) and the Cu(PrP91–115)2 complex formed in 10 mM acetate, pH 5.5, are indicated (100 μM PrP91–115, 1 mM CuSO4). The spectrum of the Cu2PrP91–115 complex formed at pH 7.4 in 50 mM Mops is also shown (60 μM PrP91–115, 200 μM copper bisglycinate).
The true dissociation constant of each peptide–copper complex is taken to be the concentration of free copper in the buffer solution when the total copper concentration is equal to the observed apparent dissociation constant. The acetate ion forms a 1:1 complex with copper (II), with a dissociation constant of 4.2 mM [36]. Since the acetate concentration is in excess of the copper concentration, the fraction of free copper in the acetate buffer can be calculated using the following relationship, which is derived from the standard binding equation for formation of a 1:1 complex:
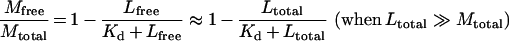 |
where Mfree is free metal concentration, Mtotal is total metal concentration, Lfree is free ligand concentration, and Ltotal is total ligand concentration.
Glycine and Tris form 2:1 and 4:1 complexes with copper (II) respectively and so a mixture of different complexes is present in each case. The calculation of the free copper concentration in these buffers is more complex and had to be carried out using the same numerical model described above. Equilibria describing the formation of the Cu(Tris)4 complex and the copper bisglycinate complex were modelled using stepwise dissociation constants from the literature [36] adjusted for pH [37]. For Tris, these constants are 0.55, 1.0, 1.7 and 4.9 mM and for glycine, they are 1.2 and 12 μM.
RESULTS
At maximum occupancy, PrP57–91 binds four copper (II) metal ions
Titration of PrP57–91 with copper (II) in NEM buffer at pH 7.4 produces a complex with strong CD bands at 350 and 580 nm (Figure 1A). The CD signal saturates at a stoichiometry of 4:1. When the signal intensity is plotted against the quantity of copper added, it can be seen that the increase does not relate linearly to the quantity of copper (Figure 1B); the change in the CD signal on adding 1 molar equivalent of copper (II) is substantially smaller than for subsequent additions. These findings are in agreement with previous studies [20,23].
Figure 1. A Cu4(PrP57–91) complex is observed using CD.

(A) Titration of 30 μM PrP57–91 with CuSO4 in 25 mM NEM buffer, pH 7.4, observed using CD, reveals a mode of copper binding which produces strong CD signals at 340 and 580 nm. Spectra are shown with 1–5 molar equivalents of copper (II) added. (B) Molar CD at 580 nm plotted against the mole fraction of copper added, showing that copper binding saturates at a stoichiometry of 4:1.
The choice of buffer is important in these experiments. Whereas NEM and other morpholine-based buffers interact only very weakly with copper (II) [37], buffers that compete more strongly for copper (II), such as Tris, will prevent formation of the CD-active complex at equivalent copper concentrations [23]. Accordingly, we have found that addition of 0.5 mM glycine in NEM buffer to the 4:1 complex at pH 7.4 abolishes approx. 80% of the CD signal (results not shown). The fact that the four copper sites in the octapeptide repeats in PrP57–91 are not maintained in the presence of glycine is consistent with the reported low affinities which are in the micromolar range [15,23]. However, the absence of a substantial CD signal after the addition of 1 molar equivalent copper (II) to the peptide in NEM buffer does not preclude the existence of a single site of higher affinity.
PrP57–91 co-ordinates a single copper (II) ion with higher affinity
Equilibrium dialysis shows that a single copper (II) ion will bind to PrP57–91 in Tris buffer at pH 7.4 (Figure 2A) with higher affinity than observed at additional sites. The apparent dissociation constant in 5 mM Tris buffer is 1.0±0.2 μM, and using the association constants for the formation of a Cu(Tris)4 complex [36], it can be calculated that 1 μM total copper (II) will give ∼3 nM free copper (II) in this buffer. Thus the dissociation constant for the Cu(PrP57–91) complex can be estimated as 3±2 nM (apparent and true dissociation constants for PrP57–91 are summarized in Table 1). Using the equilibrium dialysis data, peptide samples can be prepared for spectroscopic analysis under conditions where the amount of bound copper is accurately known. In this case, the 1:1 complex was found to produce virtually no CD signal at visible wavelengths (Figure 2C), and the form of co-ordination present can therefore not resemble that observed at maximum occupancy. The data also show that further binding can occur in these conditions (Figure 2A), but with a substantially lower affinity.
Figure 2. A 1:1 Cu(PrP57–91) complex is revealed by equilibrium dialysis with a mode of binding distinct from that in the Cu4(PrP57–91) complex.

Equilibrium dialysis of PrP57–91 shows that the peptide binds a single copper (II) ion with greater affinity than subsequent ones. Scatchard plots show the fractional occupancy (v) against v/[Cufree], with the slope of the fitted lines giving the apparent association constant and the intercept on the x-axis giving the number of sites of this affinity. (A) Dialysis against CuSO4 in 5 mM Tris buffer, pH 7.4, reveals a single high-affinity site (intercept=0.86) with an apparent dissociation constant of 1.0±0.2 μM. Binding at a second site was observed under these conditions, but is substantially weaker. (B) A single-high affinity site (intercept=1.03) is also present at pH 5.5 in 10 mM acetate buffer, with an apparent dissociation constant of 13±2 μM. (C) CD spectra show that these 1:1 complexes involve a different form of co-ordination to that seen in the 4:1 complex. The spectra of the 1:1 complexes formed in 5 mM Tris buffer at pH 7.4 (15 μM PrP57–91 and 25 μM CuSO4, i.e. 1:1 CuSO4 and 10 μM free CuSO4) and in 10 mM acetate of PrP57–91 pH 5.5 (42 μM PrP57–91 and 300 μM CuSO4, i.e. 1:1 CuSO4 and 240 μM free CuSO4) do not give any measurable CD bands, in contrast with the 4:1 complex formed in NEM buffer at pH 7.4.
Table 1. Apparent and real dissociation constants for PrP57–91.
| pH | Buffer | Number of sites | Kd (app) | Kd (real) |
|---|---|---|---|---|
| 7.4 | 5 mM Tris | 1 | 1.0±0.2 μM | 3±2 nM |
| 5.5 | 10 mM Acetate | 1 | 13±2 μM | 4±2 μM |
| 7.4 | 50 mM Mops | 2 | 6±2 μM | 250±50 nM |
A single copper (II)-binding site was also detected in acetate buffer at pH 5.5 (Figure 2B). In 10 mM acetate, the apparent dissociation constant is 13±2 μM, and from this the dissociation constant of the Cu(PrP57–91) at pH 5.5 can be calculated as 4±2 μM. This 1:1 complex also has no detectable CD signal at 580 nm (Figure 2C), suggesting a mode of metal co-ordination indistinguishable from that observed at pH 7.4.
The PrP57–91 peptide forms a 1:1 complex with copper (II) in which all four histidine residues are co-ordinated
To investigate the nature of the 1:1 complex, a sample of PrP57–91 was titrated with copper (II) and observed by one-dimensional 1H-NMR. Copper (II) is paramagnetic and will cause NMR signals from nearby nuclei to broaden to the point of becoming undetectable. The addition of copper caused specific simultaneous broadening of all histidine imidazole resonances in PrP57–91 (Figure 3). This indicates that all four histidine side chains in the peptide are involved in co-ordinating the first copper (II) ion. Co-ordination by four imidazoles is consistent with the observed 1:1 stoichiometry of the highest-affinity binding event. The CD response arises from copper (II) co-ordination in a chiral environment, which, in this case, occurs predominantly due to the chiral Cα centre between the co-ordinating imidazole and backbone amide of the histidine residue. In a multiple-imidazole-co-ordinated species, the metal ion would be substantially further from this chiral centre compared with co-ordination through both the imidazole and the backbone amide. Thus the weak CD signal observed for the 1:1 complex is also consistent with multiple histidine co-ordination. Finally, imidazole-only co-ordination would be substantially less pH-sensitive than backbone amide co-ordination owing to the lower pKa of the imidazole group. If the pH is below the pKa of a co-ordinating group in a metal-binding site, the affinity of the site is reduced by a factor of ten for each pH unit below the pKa. Since the pKa of the imidazole group is approx. 6, and the site is believed to contain four imidazole groups, the affinity of the complex would be expected to be reduced by a factor of about a hundred [i.e. 104×(6.0−5.5)] at pH 5.5 relative to pH 7.4. The observed difference is in fact ∼1000-fold, suggesting that other factors may also be involved.
Figure 3. One-dimensional NMR of the Cu(PrP57–91) complex.

The Cu(PrP57–91) complex can be observed by one-dimensional 1H-NMR. The given region of the spectra shows the aromatic histidine and tryptophan signals for a titration of 30 μM peptide with copper bisglycinate, in 25 mM phosphate buffer, pH 7.4. The mole fraction of copper added is shown on the left. Signals from the four histidine residues in the octapeptide repeats are overlapped so only combined signals are observable. Addition of copper causes specific broadening of the two histidine imidazole signals, with reduced broadening observed for the tryptophan indole signals.
Low copper occupancy causes self-association of PrP57–91
Although the 1:1 complex does not produce CD bands at visible wavelengths, it is readily observed by a reduction in the intrinsic tryptophan fluorescence of the peptide (Figure 4). We also found that the relative reduction in the fluorescence of PrP57–91 that occurs upon titration with copper is dependent on the peptide concentration. The greater degree of fluorescence quenching that occurs at higher peptide concentrations is consistent with a greater fraction of peptide being held in close proximity to a copper (II) ion. The peptide concentration affects both the rate at which fluorescence is quenched by addition of copper, and the fluorescence at the endpoint of the titration, where all the peptide should be in the 1:1 complex, Cu(PrP57–91). A simple explanation for this is that Cu(PrP57–91) can associate with other molecules of apo-peptide, at low copper concentrations, or with other Cu(PrP57–91) complexes. Since the changes in the fluorescence-quenching behaviour are seen on increasing the peptide concentration from 30 nM to 1 μM, the apparent dissociation constant for the self-association process will be within this range. It is likely that copper (II) will dissociate from these self-associated peptide complexes more slowly than from the monomeric complex. If this is the case, the self-association process will enhance the apparent affinity of copper (II) binding in the 1:1 complex. Indeed, at 30 nM peptide, the titration data show half maximal binding at a copper concentration of approx. 4 μM, whereas, at a peptide concentration of 1 μM, it was 1.5 μM.
Figure 4. Self-association of the Cu(PrP57–91) complex is demonstrated by fluorescence quenching.

The quenching of the intrinsic fluorescence, which occurs on formation of 1:1 complexes of copper and PrP57–91 in 5 mM Tris buffer, pH 7.4, is dependent on peptide concentration. Samples of PrP57–91 were titrated with copper, with peptide concentrations of 3 μM (○), 1 μM ( ), 0.3 μM (□), 0.1 μM (■) and 0.03 μM (△).
), 0.3 μM (□), 0.1 μM (■) and 0.03 μM (△).
PrP57–91 can co-ordinate two copper (II) ions with intermediate affinity
Equilibrium dialysis carried out in Mops buffer at pH 7.4, where the copper was presented as a glycine chelate, revealed a clear 2:1 stoichiometry (Figure 5A). In this case, the two sites have identical apparent dissociation constants of 6±2 μM and, given stepwise dissociation constants for copper bisglycinate of 1.2 μM and 12 μM [37], a true dissociation constant of 250±50 nM. In contrast with the 1:1 complex formed in Tris buffer, the CD spectrum of the 1:1 complex formed in this buffer had clear bands in the visible region at 350 and 580 nm (Figure 5C), indicating a different mode of copper (II) co-ordination.
Figure 5. A Cu2(PrP57–91) complex with a mode of binding distinct from that of both the 1:1 and 4:1 complexes.

In Mops buffer, with copper presented as a glycine chelate, the high-affinity 1:1 complex of PrP57–91 is disfavoured and two identical sites are observed. (A) Scatchard plot of an equilibrium dialysis experiment in 50 mM Mops buffer at pH 7.4, shows two sites (intercept=1.94) of equal affinity, each with an apparent dissociation constant of 6±1 μM. (B) Titration with Mops causes changes in the chemical shift of signals from the PrP57–91 peptide, indicating a weak interaction with the peptide. The change in the chemical shift of the histidine Hϵ1 signal of PrP57–91 is shown. To eliminate the possibility that pH effects were responsible for these changes the experiments were carried out in phosphate buffer at two different concentrations: 25 mM (○) and 50 mM (●). (C) The mode of copper binding observed in Mops buffer gave a clear CD spectrum even at 1:1 stoichiometry. Spectra were recorded for an 11 μM sample of peptide in 50 mM Mops and 10 μM glycine, pH 7.4, with one copper (II) ion bound (1:1 CuSO4 and 10 μM free copper bisglycinate), and with two copper (II) ions bound (2:1 CuSO4 and 100 μM free copper bisglycinate). Addition of copper bisglycinate to 300 μM produced no further change in the spectra, indicating that binding was saturated under these conditions at a stoichiometry of 2:1. The CD bands produced by the complexes formed in Mops buffer were similar to those observed in the 4:1 complex formed in NEM buffer at pH 7.4.
We judged that the most probable cause of the alternative copper co-ordination in this 1:1 complex was an interaction between the peptide and the Mops buffer which interferes with copper co-ordination; we had observed previously that the peptide is insoluble in the presence of Mes (chemically similar to Mops) at pH 5.5, whereas it is soluble in other buffers at the same pH (results not shown). To test the proposal that the peptide interacts with Mops, a sample of the peptide was titrated with Mops buffer at pH 7.4 and observed by NMR. Chemical-shift changes were indeed observed for the aromatic histidine signals (Figure 5B), confirming that an interaction does occur and that the bound species is significantly occupied at a Mops concentration of 50 mM, equivalent to that present in the equilibrium dialysis and CD experiments described above.
The CD spectrum of the 2:1 complex formed in Mops buffer has peaks at the same wavelengths as the 1:1 complex, but with twice the intensity, again implying that the two sites are identical. Interestingly, the positions of these CD bands are very similar to those seen previously for the Cu4(PrP57–91) complex in NEM buffer at pH 7.4. The relative signal intensity of the Cu2(PrP57–91) complex in Mops is also comparable with that seen for the Cu4(PrP57–91) complex, suggesting a similar mode of metal co-ordination. However, the metal co-ordination cannot be identical, since binding of PrP57–91 in Mops saturates at a stoichiometry of 2:1 rather than 4:1 and the two sites are of sufficiently high affinity to form in the presence of glycine.
Simulations of copper binding to PrP57–91 with three modes of co-ordination
To test the counterintuitive conclusion that, when the two forms of binding are mutually exclusive, four weak copper (II)-binding sites can out-compete a single higher-affinity site, we have used our experimentally derived dissociation constants to simulate the populations of the different species over a range of total copper (II) concentrations (Figure 6). The following equilibrium was modelled, where three mutually exclusive modes of binding are possible, which here are labelled A, B and C:
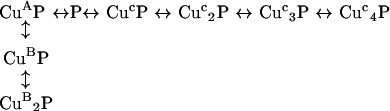 |
(2) |
A dissociation constant of 3 nM was used for the single site present in mode A and a microscopic dissociation constant of 250 nM for each of the two independent sites available in mode B, both based on our experimental measurements. A microscopic dissociation constant of 1 μM was used for each of the four independent sites available in mode C, based on literature reports of the affinity of this mode of binding [32]. The total peptide concentration was set to 30 μM to be equivalent to the CD experiments shown in Figure 1. With 1 molar equivalent of Cu2+, 94% of the peptide was in the high-affinity CuAP complex, which represents the multiple-histidine-co-ordinated complex that we have described. When the total copper concentration was increased, the higher-stoichiometry lower-affinity binding becomes dominant; with 5 molar equivalents of Cu2+ present, 97% of the peptide was in either the CuC3P or the CuC4P form, which represent complexes where each copper ion is co-ordinated by deprotonated backbone amides and a single histidine imidazole. The model therefore supports the assertion that four binding sites of micromolar affinity can out-compete a single site of nanomolar affinity. Furthermore, if the presence of the mode A species was not taken into account, the populations of the mode C species would appear to show co-operative binding. The population of the two intermediate-affinity sites (mode B: CuBP, CuB2P), with 250 nM affinity, does not exceed 15% of the total peptide concentration at any point. It is possible that the intermediate-affinity form of binding (mode B) is stronger than we have estimated. If the interaction of the peptide with the Mops buffer competes against this mode of binding, then the dissociation constant will be lower than 250 nM in the absence of Mops.
At maximum occupancy, PrP91–115 can bind two copper (II) ions
A similar program of experiments to that described for PrP57–91 was carried out on the peptide PrP91–115 to determine whether this peptide could also access multiple copper (II)-binding modes. Equilibrium dialysis data for PrP91–115 in Mops buffer at pH 7.4 with copper presented as a glycine chelate indicate that two sites of equivalent affinity were present (Figure 7A) with apparent dissociation constants of 6±1 μM and true dissociation constants of 250±30 nM. The CD spectrum of the Cu2(PrP91–115) complex which forms in Mops buffer (Figure 7D) was very similar to that reported by Jones et al. [26] with a large positive band at approx. 500 nm. It has been demonstrated by others that this complex comprises two copper-binding sites, each involving co-ordination by a single histidine imidazole, from either His96 or His111, and nearby backbone amide groups [21,26].
PrP91–115 will form a Cu(PrP91–115)2 complex
We investigated the binding interaction of PrP91–115 with copper in Tris buffer at pH 7.4 using equilibrium dialysis, and extrapolation from the first three points of the Scatchard plot shows that the highest-affinity interaction has a stoichiometry of 0.5:1 (Figure 7B). This indicates that, at low copper concentrations, each copper ion forms a complex with two peptide molecules. The equations describing the Scatchard plot for formation of such a species are non-linear. In order to estimate values for the dissociation constants describing the formation of the Cu(PrP91–115)2 species, the following equilibrium was modelled:
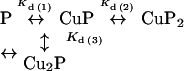 |
(3) |
Using this model, values for the dissociation constants could be fitted to the Scatchard plot, and thus Kd(1) was estimated as 30±10 μM, Kd(2) as 5±1 μM and Kd(3) as 40±10 μM. As the Kd(1) and Kd(3) values describe the binding of free copper to the peptide, these values are apparent dissociation constants. The true values were then calculated as 100±50 nM and 140±50 nM respectively (apparent and true dissociation constants for PrP91–115 are summarized in Table 2). The fact that different complexes were again observed in Tris and Mops buffers points to an interaction between the Mops buffer and the peptide that competes against formation of the Cu(PrP91–115)2 complex.
Table 2. Apparent and real dissociation constants for PrP91–115.
| pH | Buffer | Kd(1) (app) | Kd(1) (real) | Kd(2) (real) | Kd(3) (app) | Kd(3) (real) |
|---|---|---|---|---|---|---|
| 7.4 | 5 mM Tris | 30±10 μM | 100±50 nM | 5±1 μM | 40±10 μM | 140±50 nM |
| 5.5 | 10 mM Acetate | 1±0.1 mM | 300±100 μM | 40±10 μM | − | − |
| 7.4 | 50 mM Mops | 6±1 μM | 250±30 nM | − | 6±1μM | 250±30 nM |
The complexes of PrP91–115 with copper were detectable by fluorescence (results not shown). The effects of titration of this peptide with copper were again dependent on the peptide concentration, supporting the other evidence for the existence of the Cu(PrP91–115)2 complex.
The mode of copper co-ordination in the Cu(PrP91–115)2 complex is pH-dependent
A Cu(PrP91–115)2 complex will also form at pH 5.5 in 10 mM acetate buffer (Figure 7C). At this pH, there was no evidence of a Cu2(PrP91–115) complex and therefore a simplified version of the above equilibrium, which did not contain the Cu2P species, was fitted to the data to give dissociation constants of 1±0.1 mM for Kd(1) and of 40±10 μM for Kd(2). The true value for Kd(1) was then calculated as 300±100 μM, some 4000-fold weaker at pH 5.5 than at pH 7.4. The amide functional group has a pKa well above 7.4, and so if two deprotonated amides and a single imidazole are involved in co-ordinating the metal ion, then the affinity at pH 5.5 would be a factor of approx. 2×104 lower than at pH 7.4 {i.e. 10[2×(7.4−5.5)]+(6.0−5.5)}. The fact that the reduction in affinity was not this great could indicate that, at pH 5.5, the dominant Cu(PrP91–115) species is a complex with co-ordination from two histidine imidazole groups. The binding of a second peptide to the Cu(PrP91–115) complex to give Cu(PrP91–115)2 was approx. 10-fold weaker at pH 5.5 than at pH 7.4; this would be consistent with co-ordination of the copper (II) ion by two imidazoles from the second peptide, since pH 5.5 is 0.5 units below the histidine imidazole pKa. Interestingly, at pH 5.5, the second peptide bound with higher affinity than the first, an observation which could be rationalized if a direct interaction between the peptides stabilized the Cu2P species.
The Cu(PrP91–115)2 complexes at pH 7.4 and pH 5.5 both produced a CD signal at visible wavelengths (Figure 7D), but the spectra are not identical, indicating that copper (II) co-ordination is affected by pH. If these spectra were normalized by the concentration of bound copper instead of by the concentration of peptide, the band at 525 nm would be similar to the bands seen in the Cu2(PrP91–115) complex. This would be consistent with some backbone amide co-ordination occurring in the Cu(PrP91–115)2 complexes.
Investigation of the Cu(PrP91–115)2 complex by NMR
In an effort to confirm which functional groups were involved in co-ordinating copper in the Cu(PrP91–115)2 complex, a titration of the peptide with copper (II) was observed by NMR, similar to that already described for PrP57–91. In this titration, signals originating from both histidine imidazole side chains were observed to broaden simultaneously as copper was added to the sample (Figure 8A), indicating that they are both involved in the Cu(PrP91–115)2 complex. The relative populations of free peptide and the different metal complexes can be predicted on the basis of the dissociation constants fitted to the equilibrium dialysis data (Figure 8B). The total integral of the Hδ2 signal did not decrease on addition of copper to the same extent as the population of free peptide. This must be because not all the histidine signals in the Cu(PrP91–115)2 complex are broadened beyond detection. This would be the case if, of the four histidine residues in the two peptides, one or two did not directly co-ordinate copper. At least two histidine signals must become undetectable in the Cu(PrP91–115)2 complex; if only one was affected, the reduction in signal in the first part of the titration would be about half that observed. In summary, in the Cu(PrP91–115)2 complex, the copper (II) ion must be co-ordinated by either two or three histidine side chains, and probably also with a contribution from deprotonated backbone amides.
Figure 8. NMR of PrP91–115 copper (II) complexes.

Formation of the Cu(PrP91–115)2 complex can be observed by one-dimensional 1H-NMR, with broadening of the Hϵ1 signals of the two histidine residues, indicating that they are both involved in copper co-ordination. (A) Spectra are shown for a titration of 100 μM PrP91–115 with copper (II), in 5 mM Tris buffer, pH 7.4, with the mole fraction of copper (II) added shown in the left. (B) The predicted fractions of free peptide (solid line) and peptide in the Cu(PrP91–115)2 complex (broken line), based on the equilibrium dialysis data, show that the Cu(PrP91–115)2 is the dominant species below 1:1 stoichiometry. The integral of the histidine Hδ2 signal (○) is plotted relative to its starting value on the same axes.
DISCUSSION
The four octapeptide repeats in the peptide PrP57–91 can bind copper in at least three different ways (Figure 9A). This has recently been reported by other investigators who have used EPR data to show that three species exist and to propose the mode of co-ordination in each [31]. Our data support their proposals and allow us to build on the work in several important ways. We have been able to directly estimate the affinity of the two higher affinity modes of copper (II) binding. We have shown that self-association of the octapeptide repeats plays a role in the highest affinity, multiple-histidine-co-ordination mode. We have also defined conditions under which the two intermediate-affinity sites are the only species present, which was reported to be an obstacle to further spectroscopic investigation of this species by Chattopadhyay et al. [31]. The CD spectra that we were able to acquire for the Cu2(PrP57–91) complex support the proposal that the copper is co-ordinated in this complex in a similar way to the Cu4(PrP57–91) complex, but with an additional axially co-ordinated histidine.
Figure 9. Multiple modes of copper binding in PrP.

(A) At least three different modes of copper (II) co-ordination occur within the octapeptide repeats at pH 7.4. Models for the structure of each mode of binding based on our data and those of others are shown, alongside estimates for the dissociation constants. The cartoons represent the peptide backbone in the peptide PrP57–91, with the histidine side chains shown as pentagons and the copper (II) ion represented by the black circle. (B) The structure for the Cu(PrP91–115) and Cu2(PrP91–115) complexes is shown as described in the literature (left). At present, there are insufficient data to propose a detailed structure for the Cu(PrP91–115)2 complex, although our NMR data indicate that either two or three of the available histidine residues are involved in co-ordinating the metal ion (right). Estimates of the dissociation constants are given for all the complexes based on our measurements with the exception of the lowest binding affinity shown in (A) which is based on reports in the literature. The cartoons represent the peptide backbone and the side chains of His96 and His111. At present, it is not possible to distinguish whether either histidine is favoured for any particular co-ordination position.
From our data, it is also clear that at least two forms of copper co-ordination occur in the peptide PrP91–115 (Figure 9B). At maximum occupancy, two sites are observed which both involve co-ordination by a histidine imidazole and deprotonated backbone amides. The affinities of these two sites are similar, indeed they could be the same within error, and are in the range 100–200 nM. The Cu(PrP91–115) complex binds a second peptide with an affinity of approx. 5 μM; an affinity this high suggests that the second peptide is acting as a bidentate ligand because a single imidazole only binds to copper with a dissociation constant of approx. 60 μM [37]. The pH-dependence of this interaction also supports the idea that two imidazoles from the second peptide co-ordinate the copper (II) ion. The interaction could take the form of axial co-ordination from two imidazole groups or could involve the displacement of some of the backbone amides that co-ordinate copper in the Cu(PrP91–115) complex.
The existence of the Cu(PrP91–115)2 complex strongly suggests that the copper ion in the Cu(PrP91–115) complex has spare co-ordination sites that can bind a second peptide molecule through its histidine residues. As such, the peptide PrP91–115 can not be considered a complete model of copper (II) binding in the full-length protein, where additional histidine side chains would be available to fill any spare co-ordination sites. Likewise, the self-associative behaviour of the Cu(PrP57–91) complex could be explained if there were spare co-ordination sites on the metal ion that allowed intermolecular complexes to form. Again, in the full-length protein, with additional histidine side chains available, extra co-ordinating groups may be involved in the complex. If this were the case, affinities in the full-length protein would be higher than those measured in the peptide models used in the present study.
Our observations of multiple copper (II) co-ordination modes in the octapeptide repeats explain discrepancies in the literature about the stoichiometry of copper (II) binding in the octapeptide repeats and in the full-length protein. As described above, most studies have reported that the four octapeptide repeats bind four copper (II) ions with micromolar affinities at pH 7.4 [15,20,23]. Likewise, a stoichiometry of 5.2±1.1 has been observed for the full-length PrP at pH 7.4 [21]. These high-occupancy-binding modes, which have affinities in the micromolar range, will only occur in the absence of competition for copper (II) from chelating agents, such as Tris or glycine. Studies that use conditions that compete against the high-occupancy copper (II)-binding modes, such as amino acid competition or gel-filtration chromatography, have reported that the four octapeptide repeats can bind only a single copper (II) ion [12,25]. Similarly, mildly acidic pH will disfavour the deprotonation of backbone amides which is required for high-occupancy binding; at pH 6.0, the full-length protein is found to bind just two copper ions [33], instead of the five or six at pH 7.4. It is now clear that these low-occupancy copper (II)-binding modes involve co-ordination from multiple histidine imidazoles, which allows the sites to be maintained at lower pH, and that they have affinities in the low-nanomolar range, which is sufficiently strong for the site to remain intact in the presence of competition from chelating agents with micromolar affinities.
The total copper concentration in serum is 16–20 μM, all of which exists as complexes, the majority with caeruloplasmin and the remainder with other proteins (including transcuprein and albumin), peptides and amino acids [34], and the concentration of free copper (i.e. an aqua complex) is therefore virtually nil. On the cell surface in brain tissue, PrP would equilibrate with the extracellular pool of copper available in the brain interstitial fluid, which is in equilibrium with the CSF (cerebrospinal fluid). In healthy subjects, the copper concentration in the CSF is of the order of 0.1 μM [38], and, again, this is expected to exist as complexes with albumin, caeruloplasmin or amino acids. Therefore, in determining whether PrP is an authentic cuproprotein, its affinity should be compared with the affinities of the other copper-binding species in the extracellular milieu, with which it must compete for copper, and not the total copper concentration. The dissociation constant of human albumin for copper (II) is approx. 10 pM [35], whereas the stepwise dissociation constants for the formation of a copper bis-histidinate complex at pH 7.4 are approx. 4 nM and 0.5 μM [37]. Histidine is present at approx. 10 μM in the CSF, and albumin is present at approx. 20 mg/dl which equates to about 3 μM. Thus both these species are well in excess of the total copper (II) concentration and have similar or higher affinities than the peptide models of the prion protein that we have studied. Our measured affinities therefore suggest that only a small fraction of PrP, if any, is likely to bind copper under normal conditions in the central nervous system. However, during neuronal depolarization, the copper concentration at the synapse increases; one estimate places this increase as approx. 15 μM, with approx. 20% of this is in complexes with dissociation constants weaker than 100 nM [39]. It is thus likely that PrP could bind copper only when levels are elevated during neuronal depolarization. Even under this circumstance, however, probably only the higher-affinity binding modes would be populated due to the excess of other species which can bind copper (II) with micromolar or higher affinities.
The present study also raises the possibility that the elevated copper levels which occur during neuronal depolarization could play a role in controlling self-association of PrP in vivo, if the effective molarity of PrP on the cell surface is higher than approx. 100 nM. Any factor that could influence the self-association of PrP in vivo is of potential importance in understanding the normal function of PrPC and its role in prion disease and thus would merit further investigation.
Acknowledgments
This work was funded by the U.K. Medical Research Council and Biotechnology and Biological Sciences Research Council. We would like to thank Ray Young for the preparations of Figures, Neil Brammal for assistance with ICP-atomic emission spectrometry, Andrea Hounslow for assistance with NMR spectroscopy and Matthew Cliff and Rosie Staniforth for critical reading of the manuscript.
References
- 1.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 2.Griffith J. S. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner S. B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 4.Stahl N., Borchelt D. R., Hsiao K., Prusiner S. B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 5.Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wüthrich K. NMR structure of the mouse prion protein domain PrP(121–321) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 6.Zahn R., Liu A. Z., Luhrs T., Riek R., von Schroetter C., Garcia F. L., Billeter M., Calzolai L., Wider G., Wüthrich K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendheim P. E., Brown H. R., Rudelli R. D., Scala L. J., Goller N. L., Wen G. Y., Kascsak R. J., Cashman N. R., Bolton D. C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42:149–156. doi: 10.1212/wnl.42.1.149. [DOI] [PubMed] [Google Scholar]
- 8.Dodelet V. C., Cashman N. R. Prion protein expression in human leukocyte differentiation. Blood. 1998;91:1556–1561. [PubMed] [Google Scholar]
- 9.Bueler H., Fischer M., Lang Y., Bluethmann H., Lipp H. P., Dearmond S. J., Prusiner S. B., Aguet M., Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface Prp Protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 10.Collinge J., Whittington M. A., Sidle K. C. L., Smith C. J., Palmer M. S., Clarke A. R., Jefferys J. G. R. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 11.Tobler I., Gaus S. E., Deboer T., Achermann P., Fischer M., Rulicke T., Moser M., Oesch B., McBride P. A., Manson J. C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 12.Hornshaw M. P., McDermott J. R., Candy J. M. Copper-binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem. Biophys. Res. Commun. 1995;207:621–629. doi: 10.1006/bbrc.1995.1233. [DOI] [PubMed] [Google Scholar]
- 13.Hornshaw M. P., McDermott J. R., Candy J. M., Lakey J. H. Copper-binding to the N-terminal tandem repeat region of mammalian and avian prion protein: structural studies using synthetic peptides. Biochem. Biophys. Res. Commun. 1995;214:993–999. doi: 10.1006/bbrc.1995.2384. [DOI] [PubMed] [Google Scholar]
- 14.Qin K. F., Yang Y., Mastrangelo P., Westaway D. Mapping Cu(II) binding sites in prion proteins by diethyl pyrocarbonate modification and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometric footprinting. J. Biol. Chem. 2002;277:1981–1990. doi: 10.1074/jbc.M108744200. [DOI] [PubMed] [Google Scholar]
- 15.Whittal R. M., Ball H. L., Cohen F. E., Burlingame A. L., Prusiner S. B., Baldwin M. A. Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometry. Protein Sci. 2000;9:332–343. doi: 10.1110/ps.9.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown D. R., Qin K. F., Herms J. W., Madlung A., Manson J., Strome R., Fraser P. E., Kruck T., von Bohlen A., Schulz-Schaeffer W., et al. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 17.Waggoner D. J., Drisaldi B., Bartnikas T. B., Casareno R. L. B., Prohaska J. R., Gitlin J. D., Harris D. A. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J. Biol. Chem. 2000;275:7455–7458. doi: 10.1074/jbc.275.11.7455. [DOI] [PubMed] [Google Scholar]
- 18.Rachidi W., Vilette D., Guiraud P., Arlotto M., Riondel J., Laude H., Lehmann S., Favier A. Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J. Biol. Chem. 2003;278:9064–9072. doi: 10.1074/jbc.M211830200. [DOI] [PubMed] [Google Scholar]
- 19.Pauly P. C., Harris D. A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 20.Aronoff-Spencer E., Burns C. S., Avdievich N. I., Gerfen G. J., Peisach J., Antholine W. E., Ball H. L., Cohen F. E., Prusiner S. B., Millhauser G. L. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry. 2000;39:13760–13771. doi: 10.1021/bi001472t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns C. S., Aronoff-Spencer E., Legname G., Prusiner S. B., Antholine W. E., Gerfen G. J., Peisach J., Millhauser G. L. Copper coordination in the full-length, recombinant prion protein. Biochemistry. 2003;42:6794–6803. doi: 10.1021/bi027138+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonomo R. P., Imperllizzeri G., Pappalardo G., Rizzarelli E., Tabbi G. Copper(II) binding modes in the prion octapeptide PHGGGWGQ: a spectroscopic and voltammetric study. Chemistry. 2000;6:4195–4202. doi: 10.1002/1521-3765(20001117)6:22<4195::aid-chem4195>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 23.Viles J. H., Cohen F. E., Prusiner S. B., Goodin D. B., Wright P. E., Dyson H. J. Copper binding to the prion protein: Structural implications of four identical cooperative binding sites. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2042–2047. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns C. S., Aronoff-Spencer E., Dunham C. M., Lario P., Avdievich N. I., Antholine W. E., Olmstead M. M., Vrielink A., Gerfen G. J., Peisach J., et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry. 2002;41:3991–4001. doi: 10.1021/bi011922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson G. S., Murray I., Hosszu L. L. P., Gibbs N., Waltho J. P., Clarke A. R., Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8531–8535. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones C. E., Klewpatinond M., Abdelraheim S. R., Brown D. R., Viles J. H. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J. Mol. Biol. 2005;346:1393–1407. doi: 10.1016/j.jmb.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 27.Miura T., Hori-i A., Mototani H., Takeuchi H. Raman spectroscopic study on the copper(II) binding mode of prion octapeptide and its pH dependence. Biochemistry. 1999;38:11560–11569. doi: 10.1021/bi9909389. [DOI] [PubMed] [Google Scholar]
- 28.Miura T., Sasaki S., Toyama A., Takeuchi H. Copper reduction by the octapeptide repeat region of prion protein: pH dependence and implications in cellular copper uptake. Biochemistry. 2005;44:8712–8720. doi: 10.1021/bi0501784. [DOI] [PubMed] [Google Scholar]
- 29.Gustiananda M., Haris P. I., Milburn P. J., Gready J. E. Copper-induced conformational change in a marsupial prion protein repeat peptide probed using FTIR spectroscopy. FEBS Lett. 2002;512:38–42. doi: 10.1016/s0014-5793(01)03298-7. [DOI] [PubMed] [Google Scholar]
- 30.Valensin D., Luczkowski M., Mancini F. M., Legowska A., Gaggelli E., Valensin G., Rolka K., Kozlowski H. The dimeric and tetrameric octarepeat fragments of prion protein behave differently to its monomeric unit. Dalton Trans. 2004:1284–1293. doi: 10.1039/b402090a. [DOI] [PubMed] [Google Scholar]
- 31.Chattopadhyay M., Walter E. D., Newell D. J., Jackson P. J., Aronoff-Spencer E., Peisach J., Gerfen G. J., Bennett B., Antholine W. E., Millhauser G. L. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 2005;127:12647–12656. doi: 10.1021/ja053254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kramer M. L., Kratzin H. D., Schmidt B., Romer A., Windl O., Liemann S., Hornemann S., Kretzschmar H. Prion protein binds copper within the physiological concentration range. J. Biol. Chem. 2001;276:16711–16719. doi: 10.1074/jbc.M006554200. [DOI] [PubMed] [Google Scholar]
- 33.Stockel J., Safar J., Wallace A. C., Cohen F. E., Prusiner S. B. Prion protein selectively binds copper(II) ions. Biochemistry. 1998;37:7185–7193. doi: 10.1021/bi972827k. [DOI] [PubMed] [Google Scholar]
- 34.Linder M. C. Biochemistry of Copper. New York: Plenum Press; 1991. Extracellular copper substituents and mammalian copper transport; pp. 73–134. [Google Scholar]
- 35.Masuoka J., Hegenauer J., Vandyke B. R., Saltman P. Intrinsic stoichiometric equilibrium-constants for the binding of zinc(II) and copper(II) to the high-affinity site of serum-albumin. J. Biol. Chem. 1993;268:21533–21537. [PubMed] [Google Scholar]
- 36.Perrin D. D. Oxford: Pergamon Press; 1979. Stability Constants of Metal–Ion Complexes. Part B: Organic Ligands, IUPAC Chemical Data Series No. 22. [Google Scholar]
- 37.Dawson R. M. C., Elliott D. C., Elliott W. H., Jones K. M. Data for Biochemical Research. Oxford: Oxford University Press; 1986. Stability constants for metal complexes; pp. 399–416. [Google Scholar]
- 38.Joergstuerenburg H., Oechsner M., Schroeder S., Kunze K. Determinants of the copper concentration in cerebrospinal fluid. J. Neurol. Neurosurg. Psychiatry. 1999;67:252–253. doi: 10.1136/jnnp.67.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopt A., Korte S., Fink H., Panne U., Niessner R., Jahn R., Kretzchmar H., Herms J. Methods for studying synaptosomal copper release. J. Neurosci. Methods. 2003;128:159–172. doi: 10.1016/s0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]