

Abstract
When the replication fork moves through the template DNA containing lesions, daughter-strand gaps are formed opposite lesion sites. These gaps are subsequently filled-in either by translesion synthesis (TLS) or by homologous recombination. RecA filaments formed within these gaps are key intermediates for both of the gap-filling pathways. For instance, Pol V, the major lesion bypass polymerase in Escherichia coli, requires a functional interaction with the tip of the RecA filament. Here, we show that all three recombination mediator proteins RecFOR are needed to build a functionally competent RecA filament that supports efficient Pol V-mediated TLS in the presence of ssDNA-binding protein (SSB). A positive contribution of RecF protein to Pol V lesion bypass is demonstrated. When Pol III and Pol V are both present, Pol III imparts a negative effect on Pol V-mediated lesion bypass that is counteracted by the combined action of RecFOR and SSB. Mutations in recF, recO or recR gene abolish induced mutagenesis in E. coli.
Keywords: competition between Pol III and Pol V, induced mutagenesis, Pol V-mediated lesion bypass, RecFOR functions
Introduction
DNA damage is continuously generated inside cells. In addition to efficient and accurate DNA repair mechanisms that act before replication to remove most lesions, cells have evolved mechanisms that allow them to tolerate lesions at the replication fork. Such tolerance mechanisms comprise two main strategies: translesion synthesis (TLS), a potentially mutagenic strategy during which specialized DNA polymerases copy the damaged template, and damage avoidance strategies that are related to homologous recombination and are therefore essentially accurate. Experimental data on the structure and dynamics of a replication fork encountering blocking lesions are still limited. The current view is that when lesions are present in the parental DNA, the replication fork keeps going leaving gaps opposite the lesion sites in both lagging and leading strands (for a recent review, see Langston and O'Donnell, 2006; Lehmann and Fuchs, 2006). It turns out that this model, which has been proposed almost four decades ago by Rupp and Howard-Flanders, (1968) has recently received renewed attention on the basis of several in vivo (Pages and Fuchs, 2003; Lopes et al, 2006) and in vitro (Heller and Marians, 2006) reports. The gaps left in the newly synthesized strands behind the moving fork are subsequently filled-in either by TLS or by a recombination-type mechanism (recF pathway). To function in TLS, Pol V requires a physical contact with the tip of a RecA filament formed on single-stranded DNA (Dutreix et al, 1989; Fujii et al, 2004; Schlacher et al, 2006). It is thus interesting to note that both gap-filling mechanisms require a common intermediate, namely a RecA-covered ssDNA filament. Despite the fact that cells contain similar amounts of ssDNA-binding protein (SSB) and RecA proteins (Karu and Belk, 1982; Williams et al, 1984), SSB binds preferentially to ssDNA as a consequence of its much higher affinity for ssDNA than RecA (Arenson et al, 1999; Ehn et al, 2001). In all species including phages, formation of a recombinase-ssDNA filament requires the function of the so-called recombination mediator protein(s) that loads the cognate recombinase protein on single-stranded DNA covered by the cognate SSB (Gasior et al, 2001). In Escherichia coli, the recFOR gene products are thought to function as mediator proteins based on the following observations: (1) strains that carry the mutant recF, recO or recR allele show a delay in SOS induction kinetics (Hegde et al, 1995; Whitby and Lloyd, 1995) and UV sensitivity, possibly reflecting the observed degradation of nascent DNA strands, that are normally prevented by RecA function (Courcelle and Hanawalt, 2003; Chow and Courcelle, 2004) (2) specific recA alleles (e.g., recA803) are able to suppress the recF, recO or recR phenotype (Wang et al, 1993). In vitro, RecA803 protein can dissociate SSB from ssDNA efficiently compared with wt RecA (Lavery and Kowalczykowski, 1992). (3) RecOR proteins are capable to mediate in vitro an exchange reaction from SSB to RecA on ssDNA (Umezu et al, 1993). A role of RecF protein in this process has only been emerging recently (see below). Decades of work on recF, recO and recR genes have shown that RecFOR proteins are intimately involved in the establishment of a RecA filament when SSB limits RecA nucleation (Madiraju et al, 1988; Sawitzke and Stahl, 1992; Clark and Sandler, 1994; Whitby and Lloyd, 1995; Kuzminov, 1999). RecA filament formation can be viewed as a dynamic process that entails two phases: a rate-limiting nucleation step, followed by a rapid extension step; although continuous assembly and disassembly of monomers occurs at both ends in the presence of ATP, the net growth of the filament proceeds in the 5′–3′ direction by virtue of a higher association rate at the 3′ extending end (Register and Griffith, 1985; Shan et al, 1997; Bork et al, 2001a; Joo et al, 2006). Both SSB and RecX proteins modulate the formation of the RecA filament. Whereas SSB proteins at sub-stoichiometric concentrations favor the RecA filament extension phase by dissolving secondary structures, stoichiometric concentrations of SSB block the RecA nucleation phase. RecOR proteins allow to overcome the SSB-mediated nucleation block. On the other hand, RecF protein specifically overcomes the effect of RecX that otherwise blocks the RecA filament extension phase (Lusetti et al, 2006).
We have previously determined the optimal conditions for Pol V-mediated bypass using single-stranded circular templates with a single lesion. The use of a circular template was found to be instrumental by supporting the stable loading of the beta-clamp and the formation of a dynamic RecA filament in the presence of ATP (Fujii et al, 2004). Under these conditions, Pol V extends the primer several nucleotides past the lesion generating a ‘TLS patch'. Synthesis of such a TLS patch by the specialized DNA polymerase in a single binding event conceals the distortion created by the lesion, thus preventing degradation of the nascent primer by the proofreading activity associated with the replicative polymerase (Fujii and Fuchs, 2004). This step is thus crucial for successful completion of the TLS pathway. Our optimal assay conditions contained a low, sub-stoichiometric amount of SSB protein that was found to stimulate the bypass reaction, most probably by virtue of its capacity to accelerate the extension of RecA filaments at positions of secondary structures (Fujii and Fuchs, 2004; Fujii et al, 2004). However, physiologically relevant concentrations of SSB totally inhibit the Pol V-mediated TLS reaction (Fujii et al, 2004). In the present paper, we show that in the presence of saturating concentrations of SSB, efficient Pol V-mediated lesion bypass is achieved in the presence of RecFOR proteins. In addition, when Pol III and Pol V are simultaneously present in the reaction mix, Pol III appears to severely hinder Pol V-mediated TLS. Addition of RecFOR and SSB proteins appears to reverse the adverse effect of Pol III on Pol V-mediated bypass. In good agreement with these in vitro data, inactivation of either recF, recO or recR gene inhibits Pol V-mediated UV-induced mutagenesis in a forward mutation assay to the same extent as a umuDC mutant.
Results
Forward mutation assay and TLS in recF, recO and recR mutant strains
In E. coli, induced mutagenesis is known to depend upon several key proteins, namely Pol V, a Y-family polymerase responsible for the bypass of most lesions, the beta-clamp, the general replication processivity factor, and RecA protein (for a review, see Fuchs et al, 2004). Using the Weigle mutagenesis protocol, it was reported that a host recF mutation severely inhibits UV and apurinic site mutagenesis (Schaaper et al, 1982; Wood and Stein, 1986). In contrast, conflicting results have been reported using different chromosomal reversion assays. Indeed, while reversion assays based on hisG4 and argE3 alleles showed that recF and recR mutants are normal with respect to UV-induced mutagenesis (Kato et al, 1977; Mahdi and Lloyd, 1989; Clark and Sandler, 1994), the reversion frequency of the trpE65 allele was found to be severely reduced in all three recF, recO and recR mutant strains (Liu et al, 1998). Here, we show that inactivation of either recF, recO or recR gene strongly reduces UV-induced mutagenesis using a chromosomal mutation assay as well as a plasmid-based assay capable of directly measuring Pol V-mediated translesion synthesis opposite a single UV-induced lesion, namely the TT(6-4) photoproduct in vivo.
UV-induced mutagenesis: rifR assay. Induction of rifampicin-resistant colonies following UV irradiation was monitored as described in Materials and methods. The frequency of induced mutations was determined at various UV doses in wild-type strain and in isogenic recF, recO and recR mutant allele strains. The wild-type strain exhibits a robust induction of rifR mutants, reaching about 3 × 10−6 at a UV dose corresponding to 1% survival. This value corresponds to ≈100-fold increase above background (≈3–5 × 10−8) (Figure 1A). For all three rec− strains, the induction of UV-induced mutants was 5- to 10-fold reduced when compared with the wild-type strain, reaching a level similar to that observed in a umuC mutant (Figure 1A).
Figure 1.
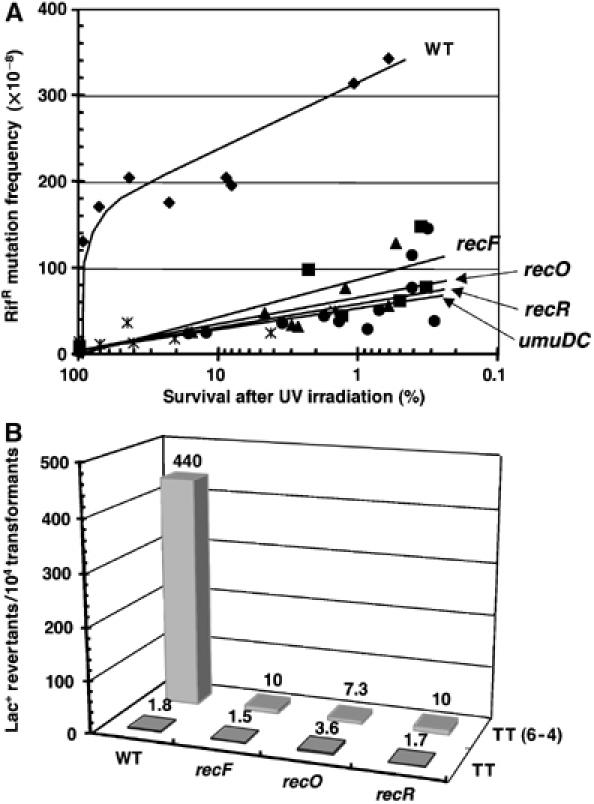
UV-induced mutagenesis. (A) Rifampicin resistance chromosomal mutagenesis assay. The frequency of UV-induced RifR colonies is plotted against survival in wild-type, recF, recO, recR and umuDC strains in the MG1655 background (see Materials and methods for additional details). Plotting mutagenesis against UV survival rather than UV dose allows one to take into account the UV sensitivity of recF, recO and recR strains. (B) Mutagenesis induced by a single TT(6-4) lesion. A single TT(6-4) photoproduct was introduced in a double-stranded plasmid vector, as reported previously (Becherel and Fuchs, 1999). The adduct is located in the lacZ (alpha) gene in a way that allows direct monitoring of mutagenic bypass by a change in colony color on lacZ indicator plate (see Materials and methods). All strains contain a low-copy number plasmid pRW134 expressing Pol V (Frank et al, 1996) at a level similar to that found in UV-induced cells. The numbers represent the average mutation frequency determined from three independent experiments.
Single TT(6-4)-induced mutation frequency. We also implemented a specific mutation assay involving a single TT(6-4) lesion located on a double-stranded plasmid vector. Both error-free and mutagenic bypass of this lesion were shown to depend entirely on Pol V (Becherel and Fuchs, 1999). The expression of Pol V calls for a functional SOS system at both the transcriptional and post-translational levels. In order to circumvent the difficulty related to the fact that recF, recO or recR strains are known to have delayed SOS induction kinetics (Hegde et al, 1995; Whitby and Lloyd, 1995) compared to a wild-type strain, we transformed our strains with a low-copy vector that constitutively expressed Pol V at a level similar to SOS-induced cells (Frank et al, 1996). We then introduced the TT(6-4) lesion-containing plasmid in these strains to score mutagenic lesion bypass directly on indicator plates as blue colonies (see Material and methods). The mutant colony fraction reached 4.4% in the wild-type strain, whereas this response was totally abolished in the recF, recO or recR strains (Figure 1B), demonstrating the direct involvement of RecFOR in Pol V-mediated TLS. In the wild-type strain, the mutagenic response is completely dependant on the presence of the Pol V-encoding plasmid (data not shown).
Reconstitution of the TLS reaction in the presence of Pol III HE, Pol V, SSB and RecA
Physiological concentrations of SSB completely block Pol V-mediated TLS: this block is relieved by the addition of RecFOR proteins. In vivo, ssDNA gaps are formed when the replicative DNA polymerase dissociates from its template, generating, in most cases, a 3′-end located at the position immediately preceding the lesion site (position L-1). To mimic the formation of such a primer by Pol III in vitro, we used a primer that ends 8 nucleotides upstream of the lesion site, allowing it to be elongated by Pol III (running start experiments). We implemented the previously optimized TLS assay conditions involving a single-stranded circular template primed with a 32P-labeled oligonucleotide in the vicinity of a single DNA lesion (G-AAF adduct) (Fujii et al, 2004). This substrate was incubated successively with purified SSB (5 min) and RecA (3 min) proteins in quantities that are sufficient for full coverage of the single-stranded DNA present in the assay. Following addition of RecFOR proteins (5 min), the substrate was incubated in the presence of Pol III HE, followed by Pol V (quantities and incubation time as indicated in Figure 2 and in Materials and methods) before the reaction is stopped for PAGE analysis.
Figure 2.
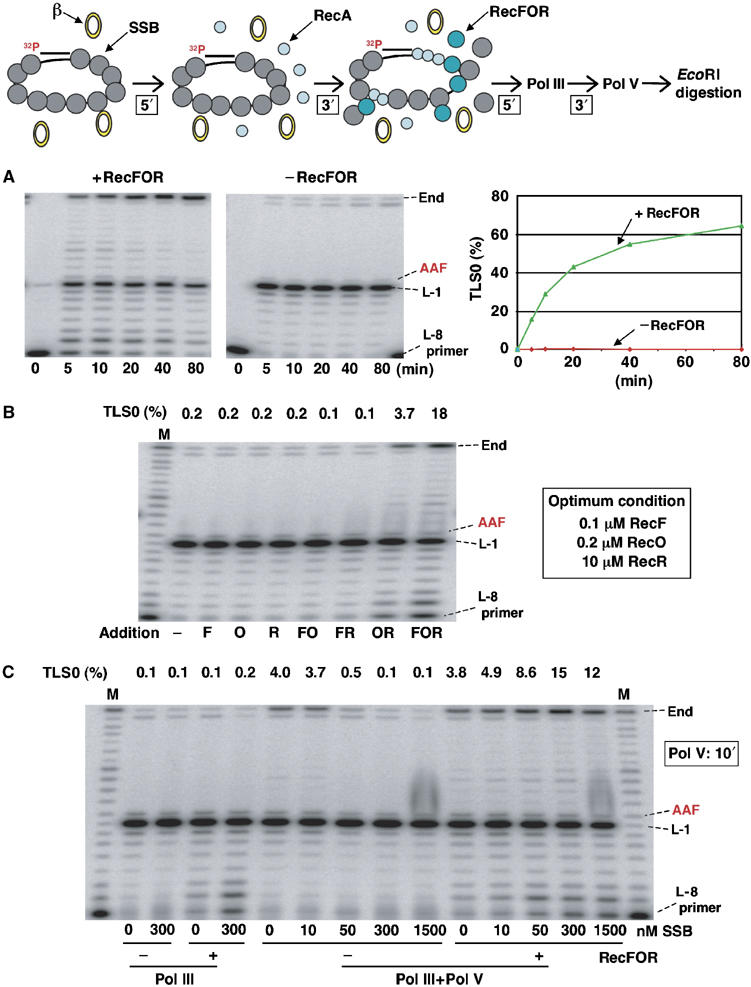
Pol III/Pol V-mediated TLS reaction in the presence of RecA and SSB proteins. (A) RecFOR proteins efficiently allow Pol III/Pol V-mediated TLS. The concentrations of RecA and SSB tetramers to fully saturate the ssDNA template present in the assay (2 nM of single-stranded circular plasmid 2.7-kb long) are 1.8 μM and 160 nM, respectively. Assays are performed at 2 μM and 300 nM of RecA and SSB tetramers, respectively. In the presence of optimal concentrations of RecFOR proteins, a robust TLS reaction is observed (>60%) at 80 min incubation. In contrast, in the absence of RecFOR proteins, essentially no bypass is observed, as the TLS0 band observed in the gel corresponds to a 0.2% contamination with lesion-free template. Percentage of TLS0 is calculated as specified in Materials and methods. (B) None of the RecF, RecO or RecR proteins alone or in combination of two have an effect on TLS, except for the RecOR combination. Further addition of RecF increases the reaction efficiency. Incubation time: Pol III, 3 min; Pol V, 10 min. (C) When both Pol III and Pol V are present, high concentrations of SSB and optimal concentrations of RecFOR proteins are necessary to yield optimal bypass conditions. In the absence of RecFOR proteins, no or low amounts of SSB protein (10 nM, i.e., 5–10% of the stochiometric amount) yield optimal bypass. However, a four-fold higher bypass reaction is observed in the presence of RecFOR and saturating amounts of SSB tetramer. Incubation time: Pol III, 3 min; Pol V, 10 min; in the absence of Pol V, incubation time with Pol III is 13 min.
The concentrations of RecF (0.1 μM), RecO (0.2 μM) and RecR (10 μM) that achieve optimal Pol V bypass have been determined by various titration experiments (see Supplementary Figures S1, S2, S4 and S5). As seen in Figure 2A, in the presence of the RecFOR proteins, a robust time-dependent appearance of full-length TLS product (TLS0) can be observed reaching ≈60% of bypass at 1 h. A strong pause site corresponding to the position preceding the lesion site (L-1) is rapidly observed and becomes less intense over time when bypass occurs. In contrast, in the absence of RecFOR proteins, the primer is rapidly elongated until position L-1, whereas essentially no TLS takes place under these conditions. Indeed, the faint full-length band at position TLS0 is constant over time and corresponds to a small contamination with non-modified substrate (≈0.2%). In contrast, the faint band at position TLS-1 slightly increases over time, and is due to a weak frameshift bypass activity mediated by Pol III HE (≈0.8% at 80 min).
In conclusion, in the presence of Pol III HE and saturating concentration of SSB and RecA, RecFOR proteins are necessary to reach high levels of Pol V-mediated lesion bypass. We suggest that RecFOR proteins are required for RecA filament formation in the presence of physiologically relevant concentrations of SSB. While under normal conditions, the cellular concentrations of RecA and SSB monomers are comparable, when SOS is induced, RecA concentration increases about 10-fold. However, given the much higher affinity for binding to ssDNA of SSB (Ehn et al, 2001) compared with RecA (Arenson et al, 1999), the competition is still largely in favor of SSB.
Pol V-mediated TLS in the presence of SSB: RecOR proteins are absolutely necessary, whereas RecF further stimulates the reaction. None of the RecF, RecO or RecR protein taken alone or in combination of two is able to stimulate TLS above background level, except for the RecOR combination (Figure 2B). Indeed, the RecOR combination allows the TLS reaction to reach a level very significantly above background (3.7% for a 10 min incubation time with Pol V). Previously published biochemical data have established that the RecOR complex functions to speed up the formation of a RecA filament on ssDNA prebound by the SSB protein (Umezu and Kolodner, 1994; Shan et al, 1997; Bork et al, 2001b). In these experiments, addition of RecF had either no or even a negative effect on the capacity of RecOR to load RecA. In contrast, we found that addition of RecF (0.1 μM) further stimulated the TLS reaction about five-fold (Figure 2B). Similarly, by monitoring the ATPase activity of the RecA filament, it was recently shown that RecF stimulates RecA filament formation on ssDNA gaps coated with SSB protein (Morimatsu and Kowalczykowski, 2003).
TLS in the presence of Pol III and Pol V polymerases: effect of SSB. We wanted to investigate the effect of SSB concentration on the efficiency of the TLS reaction in the presence and absence of RecFOR proteins. In the absence of RecFOR proteins, low concentrations of SSB (10 nM, i.e., 5–10% of the stoichiometric amount) stimulated Pol V activity by promoting RecA filament extension, whereas increasing amounts of SSB (i.e., up to stoichiometric amounts) totally inhibited the bypass reaction, as shown previously (Fujii et al, 2004). This observation reflects the two aspects of SSB action: although SSB strongly inhibits the nucleation phase of RecA filament assembly, it stimulates the extension phase by dissolving the secondary structures in ssDNA that would otherwise impede filament growth (Kowalczykowski and Krupp, 1987; Kowalczykowski et al, 1987; Lavery and Kowalczykowski, 1992). Surprisingly, in the presence of RecFOR proteins, increasing amounts of SSB very efficiently stimulated the bypass reaction, reaching levels that are in fact higher than in the absence of SSB and RecFOR proteins (Figure 2C). Kinetics of lesion bypass experiments were performed to further investigate the nature of this effect (see below and Figure 4).
Figure 4.
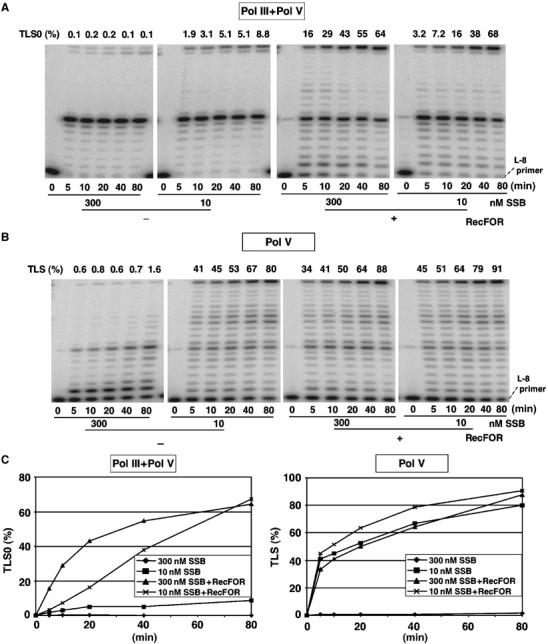
Kinetics of TLS as a function of SSB and RecFOR concentrations. (A) Pol III/Pol V-mediated TLS: in the presence of RecFOR, the bypass reaction kinetics is faster with saturating amounts of SSB compared with low amounts of SSB, whereas similar levels are reached at 80 min. In the absence of RecFOR, saturating amounts of SSB completely inhibit the reaction, whereas low amounts of SSB yield an ≈10-fold reduced level of bypass compared with the +RecFOR conditions. (B) Pol V alone (no Pol III): in order to load the beta-clamp, we added purified gamma-complex clamp loader at 10 nM final concentration. In the presence of RecFOR, the bypass reaction is efficient with either low or high amounts of SSB. In the absence of RecFOR, the bypass reaction is efficient at low SSB concentrations, but is completely inhibited at high SSB concentrations. Under these conditions, the bypass reaction merely reflects the efficiency of RecA filament formation. (C) TLS0 kinetic curves under the conditions shown in (A, B).
Role of the 5′-primer end (matched versus tailed) in RecFOR stimulation of Pol V-mediated TLS
Using the ATPase activity as a reporter for RecA filament formation, the presence of a perfectly matched 5′-ds/ss junction was reported to be important for RecF-mediated stimulation of RecA filament formation on SSB-coated ssDNA gaps (Morimatsu and Kowalczykowski, 2003). Indeed, the presence of a 5′-unpaired region (5′-tail) was shown to abolish RecF stimulation (Morimatsu and Kowalczykowski, 2003). We wanted to investigate the effect of a 5′-tail on Pol V-mediated TLS. Compared to a perfectly matched 5′-ds/ss junction, we observed a three-fold reduction with the 5′-tailed structure (compare Figures 2B and 3A). However, we cannot attribute this reduction specifically to RecF, as a similar reduction in the stimulation was observed with RecOR alone (Figure 3B). In fact, with the tailed substrate, when compared to RecOR alone, addition of RecF stimulated very significantly the kinetics of TLS (Figure 3B).
Figure 3.
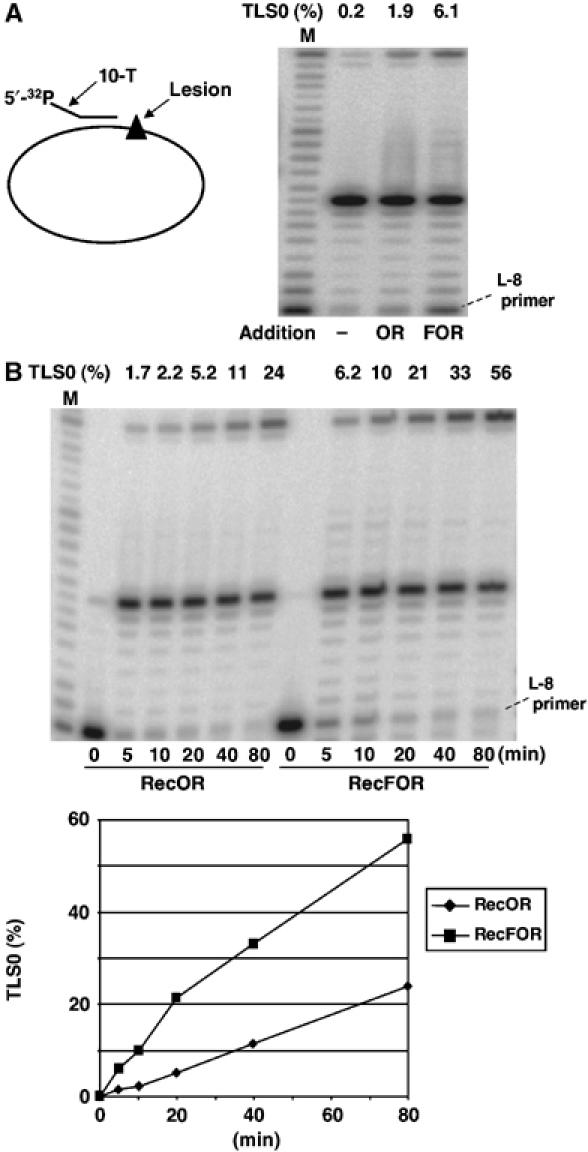
Effect of 5′-tailed primer on RecFOR stimulation of Pol V-mediated bypass. A primer containing 10 dT residues at its 5′-end was used. Incubation time: Pol III, 3 min; Pol V, 10 min. (A) A three-fold reduction in lesion bypass was observed when compared to the fully matched primer (see Figure 2B). (B) Despite an overall reduction in lesion bypass with the tailed versus the matched primer, RecF clearly stimulated the bypass reaction observed in the presence of RecOR, as seen with the perfectly paired primer (see Figure 2B).
Pol V-mediated bypass reaction is severely inhibited by the presence of Pol III HE: RecFOR and SSB proteins can reverse the adverse effect of Pol III
Using concentrations of RecA (2 μM) and SSB (10 nM) previously determined for optimal Pol V-mediated bypass, we observed that the presence of Pol III HE in the reaction mixture severely decreases (≈10-fold decrease) the kinetics and extent of the TLS reaction (Figure 4). Addition of RecFOR proteins allowed the TLS reaction to recover to levels similar to those observed in the absence of Pol III HE (Figure 4). However, a distinct effect was observed in terms of kinetics depending on SSB concentration. At low SSB concentrations, we observed full recovery of TLS but with a reduced kinetics, whereas at high SSB concentrations, the kinetics of the TLS reaction was similar to that observed in the absence of Pol III (Figure 4). How can these effects be explained? Compared with the situation with Pol V alone, when Pol III is present, it may interfere with the Pol V-mediated bypass reaction by competing for primer template binding at position L-1 (Figure 6A). As binding of Pol III is non-productive for lesion bypass, Pol III may be expected to interfere with Pol V binding. However, the large decrease in TLS efficiency is not compatible with a simple model of competition for substrate binding. Indeed, at constant Pol V concentration (100 nM), a large increase in Pol III concentration, from 4 to 60 nM, only further reduced TLS efficiency by a factor of two (data not shown). We hypothesized that binding of Pol III actually disturbs the 3′-tip of the RecA filament that is essential for Pol V bypass. Indeed, it is unlikely that binding of Pol III to sites within the RecA filament that are distant from its lesion proximal 3′-tip would influence Pol V-mediated TLS activity. Therefore, RecFOR proteins together with SSB appear to stimulate the TLS reaction by allowing proper and rapid reconstruction of the tip of the RecA filament that is perturbed by (repeated) binding of Pol III HE (Figure 6A). The precise mechanism of RecA filament reconstruction is presently unknown.
Figure 6.
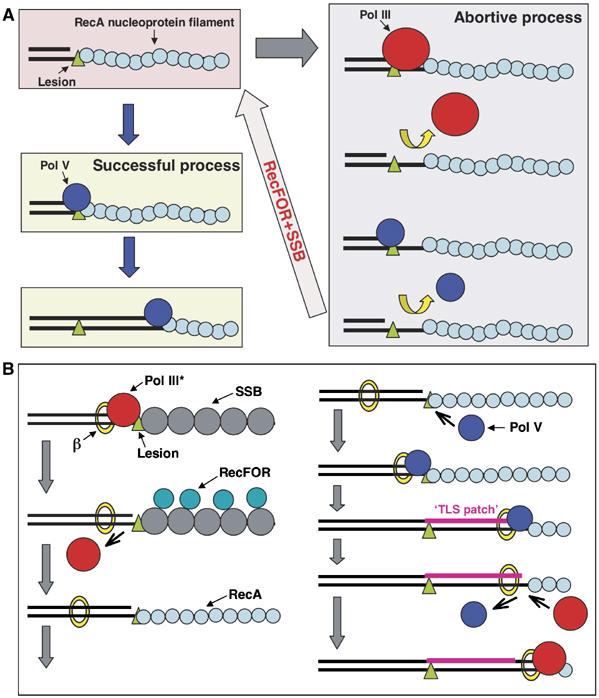
Model for TLS. (A) Interference between Pol III and Pol V. The results presented in this paper suggest that when a RecA/ATP filament is formed along the single-stranded DNA located downstream of a replication blocking lesion, the 3′-OH end of the primer can bind either Pol V or Pol III. When Pol V binds, TLS proceeds successfully as described previously (Fujii and Fuchs, 2004; Fujii et al, 2004). In contrast, binding of Pol III is not only an abortive event for the TLS reaction per se, but it also appears to locally perturb the structure of the 3′-tip of the RecA filament, thereby preventing successful binding of Pol V. Interestingly, our results show that both RecFOR and SSB proteins are able to rapidly ‘reconstruct' the RecA filament for efficient Pol V-mediated lesion bypass, thus counteracting the adverse effect of Pol III on the bypass reaction. (B) Integrated TLS model: this scheme brings together the current data for the bypass of lesion by Pol V in E. coli. Upon reaching a replication block, Pol III dissociates leaving the beta-clamp on the template, while the replication fork helicase keeps moving, thus creating a region of single-stranded DNA downstream of the lesion (Higuchi et al, 2003; Pages and Fuchs, 2003; McInerney and O'Donnell, 2004). Owing to the higher affinity for ssDNA of SSB compared with RecA, SSB will bind first, thus preventing RecA filament formation. The RecFOR mediator proteins are essential for the formation of a RecA filament. Upon binding to the 3′-OH of the primer, Pol V will form a relatively stable initiation complex via a dual interaction with the beta-clamp and the tip of the RecA filament. Synthesis of a ‘TLS patch' in excess of five nucleotides will allow Pol III to resume synthesis. We envision the process of TLS as a gap-filling reaction that occurs ‘behind' the replication fork possibly involving free Pol III core rather than the Pol III holoenzyme (Lehmann and Fuchs, 2006).
Bypass of the major UV-induced lesions: TT cyclobutane dimer (CPD) and TT(6-4) photoproduct
Experiments similar to those conducted with the single G-AAF adduct have been performed with substrates containing either a single TT CPD or TT(6-4) lesion (Figure 5). In the absence of RecFOR proteins, no bypass is seen, as the weak band at the top of the gel represents a low fraction (≈2%) of lesion-free template contained in our construction. After correction for this contamination, a comparable time-dependent accumulation of full-length TLS product is observed for both UV-induced lesions. Under optimal conditions, bypass of all three lesions is comparable with the following slight differences in efficiencies: G-AAF>TT(6-4)>TT CPD.
Figure 5.
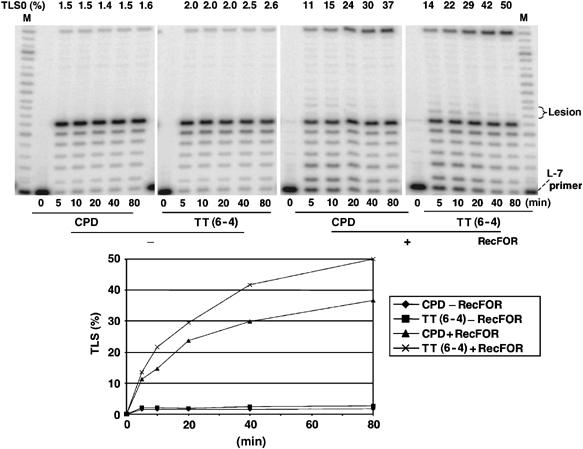
Lesion bypass of UV-induced TT lesions. The reaction conditions are the same as described for the G-AAF bypass in Figure 4A, except for the concentration of Pol III (96 nM instead of 62.7 nM) with saturating amounts of SSB (300 nM). Robust bypass is observed for both TT CPD and TT(6-4) lesions when RecFOR proteins are present. No bypass is observed in the absence of RecFOR; the weak TLS0 band is due to a low (≈2%) contamination by lesion-free template.
Discussion and conclusion
In vivo, SSB and RecA are abundant proteins that exhibit affinity for single-stranded DNA. Given the much higher affinity for ssDNA of SSB compared with RecA (Arenson et al, 1999; Ehn et al, 2001), ssDNA is preferentially covered by SSB, preventing RecA filament formation and thus inhibiting RecA filament-mediated cellular functions. A scenario similar to the one seen in E. coli seen in most organisms: a given recombinase (RecA) requires the assistance of recombination mediator proteins (RecFOR) to dislodge the cognate SSB from ssDNA. Here, we demonstrate that RecFOR proteins are essential for efficient Pol V-mediated lesion bypass under conditions that mimic the in vivo situation, that is, in the presence of RecA and SSB proteins at concentrations that fully saturate the ssDNA substrate. None of the RecF, RecO or RecR protein alone or in any combination of two is able to activate Pol V-mediated TLS, except for the RecO plus RecR combination (Figure 2B). Addition of RecF to the RecOR combination further stimulates the TLS reaction, reaching an optimum at 0.1 μM of RecF (Supplementary Figure S4). We suggest that RecFOR proteins trigger lesion bypass by allowing efficient formation of a RecA filament that is an essential cofactor for Pol V's TLS activity. The precise role of RecA in Pol V activation is still a matter of debate (see Pham et al, 2001; Reuven et al, 2001; Fuchs et al, 2004; Fujii et al, 2004; Schlacher et al, 2005). Recently, using short oligonucleotide templates, Goodman and co-workers suggested that efficient TLS requires activation of Pol V by a RecA filament in trans (Schlacher et al, 2006). However, in our hands, using a long single-stranded circular template, efficient Pol V lesion bypass activity does not rely upon the addition of a RecA filament in trans (Fujii et al, 2004).
The effect of RecFOR proteins on RecA filament formation in the presence of saturating amounts of SSB can be monitored by several assays: (1) an ATPase assay specific for the ssDNA.RecA complex; (2) a LexA or UmuD cleavage assay; (3) a homologous recombination assay based on joint molecule formation; and (4) the present assay monitoring Pol V-mediated TLS. While Pol V activation for TLS is thought to be specifically mediated by the 3′-tip of the filament (Fujii et al, 2004; Schlacher et al, 2006), all the other assays monitor the ‘internal portions' of the RecA filament instead. For all RecA filament formation assays in the presence of SSB, the RecOR complex is found to be either essential or to strongly speed up the reaction, whereas RecF acts as a modulator, stimulating or inhibiting the reactions depending on its concentration (see Supplementary Figure S2; Umezu et al, 1993; Shan et al, 1997; Xu and Marians, 2003). The role of RecF as a modulator rather than a core component of the recombination mediator apparatus is supported in vivo by the fact that recF phenotypes can partially be suppressed by RecR overexpression (Sandler and Clark, 1994) or by disruption of the recX gene (Lusetti et al, 2006).
How can we envision the role of RecFOR in RecA filament formation? RecA filament formation entails two basic steps: a rate-limiting nucleation step, followed by a rapid extension step. The RecOR complex by interacting with SSB.ssDNA (Griffin and Kolodner, 1990; Luisi-DeLuca and Kolodner, 1994) may displace the SSB filament, thus providing space for the RecA nucleation step. We envision that RecF either enhances RecOR function or assists the RecA nucleation step. In most in vitro assays published so far, RecF was reported to have either no or an inhibitory effect, except for two recent reports: (1) RecF specifically allows RecFOR to bind the ss/ds DNA junction at a fully base-paired 5′-terminus (Morimatsu and Kowalczykowski, 2003) and (2) RecF protein physically binds to RecX protein and antagonizes the effect of RecX that otherwise inhibits RecA filament formation (Lusetti et al, 2006). We show an additional property for RecF, namely its capacity to stimulate PolV-mediated bypass (Figures 2 and 3) independently of the presence of a fully paired ss/ds 5′-junction suggesting that the nucleation step does not require such a junction but can take place at any place along the ssDNA stretch instead. Alternatively, a direct role of RecF in Pol V-mediated TLS cannot be excluded.
On the basis of the biochemical data presented here and previously (Fujii and Fuchs, 2004; Fujii et al, 2004), we propose an integrated model for lesion bypass (Figure 6B) that takes into account all known genetic requirements for induced mutagenesis. In E. coli, induced mutagenesis depends upon several key gene products (for recent reviews, see Goodman, 2002; Fuchs et al, 2004; Friedberg et al, 2005), namely umuDC-(Kato et al, 1977; Steinborn, 1978) encoding Pol V, a Y-family polymerase responsible for the bypass of most lesions, dnaN, encoding the beta-clamp the general replication processivity factor (Becherel et al, 2002) and recA (Dutreix et al, 1989; Sweasy et al, 1990; Bailone et al, 1991). In addition to these factors, this paper illustrates the key function of recF, recO and recR genes in Pol V-mediated lesion bypass. We specifically show that the defect in Pol V-mediated lesion bypass in recF, recO and recR strains is not due to the known delay in SOS induction kinetics in these strains (Hegde et al, 1995; Whitby and Lloyd, 1995). Indeed, the role of RecFOR proteins in Pol V-mediated lesion bypass directly hinges on the capacity of these proteins to load a RecA filament on single-stranded DNA coated by SSB. These RecA-covered regions appear to represent common intermediates for two alternative gap-filling pathways, namely Pol V-mediated TLS or recF-mediated recombination. Further experiments are required to shed light on the mechanisms that control the respective use of these two pathways. In eukaryotes, although the direct role of a recombination filament has not yet been proven to be essential in lesion bypass, it was recently reported that RAD51 interacts with Pol eta and stimulates Pol eta-mediated D-loop extension (McIlwraith et al, 2005).
Materials and methods
Strains and mutation assays
Strains. Isogenic recF, recO and recR strains were constructed in the MGZ strain background derived from MG1655 (Napolitano et al, 2000) by P1 transduction using the following donor strains: [recF400∷Tn5] from strain JJC450, [recD1009 recO∷Tn5] from strain JJC404 and [recR∷Tn5] from strain JJC1193. The following concentrations of antibiotics were used: 10 μg/ml tetracycline and 20 μg/ml kanamycin.
Survival curves. UV survival curves were established starting from fresh overnight cultures diluted 1/50 and grown under aeration to OD700=0.5. Cells were pelleted and resuspended in 0.5 volume of 10 mM MgSO4. Aliquots of 10 ml were irradiated with 254 nm UV light at doses ranging from 0 to 60 J/m2. Adequate dilutions were plated on LB medium covered by aluminum foil to prevent potential photoreactivation.
Rifampicin resistance mutation assay
Cell irradiated at various UV doses were inoculated in LB medium (1/20 dilution) and shaken for 4 h at 37°C (0.5 ml suspended cells+9.5 ml LB). Following this period, the cultures were centrifuged, resuspended and plated on either rifampicin plates (2–4 plates, rif=100 μg/ml) or LB plates for determining the mutant and surviving fractions, respectively.
Single-adduct plasmid mutation assay
The different strains, the wild-type MGZ strain and the corresponding recF, recO and recR mutant strains were transformed with the low-copy number plasmid expressing Pol V from its wild-type promoter pRW134 (=O+-umuD′C) or the corresponding empty vector (pGB2). The corresponding strains were subsequently transformed with pCU-derived plasmids containing a single TT(6-4) photoproduct (Becherel and Fuchs, 1999) or the corresponding lesion-free plasmid and plated on indicator plates including ampicillin (Ap), spectinomycin (Spc), IPTG (0.3 mM) and X-gal (60 μg/ml). The photoproduct is located close to the 5′-end of the LacZ (alpha) gene. As the 3′-T of the photoproduct is part of an in-frame TAA ochre codon, mutagenic bypass can be monitored directly on these plates as a change in colony color from white to blue. Indeed, the most frequent mutagenic bypass event of the 6-4 photoproduct, a T → C transition at 3′-T, converts the ochre codon into a sense codon (Becherel and Fuchs, 1999).
Proteins
PoI III*, Pol V, the beta-clamp and SSB were prepared as described (Fujii and Fuchs, 2004; Fujii et al, 2004). RecA was obtained from Pharmacia. The gamma-complex clamp loader and RecFOR proteins were generous gifts from CS McHenry (University of Colorado, Denver) and from KJ Marians (Memorial Sloan-Kettering Cancer Center, NY), respectively. DNA substrates are described in the figure legends and elsewhere (Fujii and Fuchs, 2004; Fujii et al, 2004).
DNA replication assays
The components of reaction buffer were 20 mM Tris–HCl (pH 7.5), 4% glycerol, 8 mM DTT, 80 μg/ml BSA, 2.5 mM ATP, 8 mM MgCl2 and 0.1 mM each dNTP (dA, dT, dG and dC). The standard reaction was carried out as follows: 300 nM SSB (as a tetramer), 50 nM beta-clamp (as a dimer) and 2 nM primed template DNA were mixed and incubated for 5 min at 30°C, followed by the addition of 2 μM RecA. After 3 min, 0.1 μM RecF, 0.2 μM RecO and 10 μM RecR were mixed and incubated for 5 min at 30°C. The reaction was initiated by the addition of 62.7 nM Pol III*. After 3 min, 100 nM Pol V was mixed and incubated for 10 min at 30°C. The reaction was terminated by adding EDTA (the final concentration was 25 mM). Thereafter, the products were purified by phenol/chloroform extraction, followed by ethanol precipitation and EcoRI digestion. The reactions were stopped by adding 1 volume of formamide containing 25 mM EDTA and bromophenol blue. The products were heat denatured, separated on a 10% denaturing PAGE and visualized on a Molecular Imager (Bio-Rad). The percentage of TLS was calculated as follows: in the presence of Pol III and Pol V, TLS0=intensity of TLS0 band/sum of TLS0+L-1+L0; with Pol V alone, TLS0=sum of all bands above L0/sum of all bands above L-8.
Supplementary Material
Supplementary Material Figures S1, S2, S4 and S5
Acknowledgments
We thank CS McHenry (University of Colorado, Denver) and KJ Marians (Memorial Sloan-Kettering Cancer Center, NY) for generous gifts of purified gamma-complex clamp loader and RecFOR proteins, respectively.
References
- Arenson TA, Tsodikov OV, Cox MM (1999) Quantitative analysis of the kinetics of end-dependent disassembly of RecA filaments from ssDNA. J Mol Biol 288: 391–401 [DOI] [PubMed] [Google Scholar]
- Bailone A, Sommer S, Knezevic J, Dutreix M, Devoret R (1991) A RecA protein mutant deficient in its interaction with the UmuDC complex. Biochimie 73: 479–484 [DOI] [PubMed] [Google Scholar]
- Becherel OJ, Fuchs RP (1999) SOS mutagenesis results from up-regulation of translesion synthesis. J Mol Biol 294: 299–306 [DOI] [PubMed] [Google Scholar]
- Becherel OJ, Fuchs RPP, Wagner J (2002) Pivotal role of the β-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair 1: 703–708 [DOI] [PubMed] [Google Scholar]
- Bork JM, Cox MM, Inman RB (2001a) RecA protein filaments disassemble in the 5′–3′ direction on single-stranded DNA. J Biol Chem 276: 45740–45743 [DOI] [PubMed] [Google Scholar]
- Bork JM, Cox MM, Inman RB (2001b) The RecOR protein's modulate RecA protein function at 5′ ends of single-stranded DNA. EMBO J 20: 7313–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KH, Courcelle J (2004) RecO acts with RecF and RecR to protect and maintain replication forks blocked by UV-induced DNA damage in Escherichia coli. J Biol Chem 279: 3492–3496 [DOI] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ (1994) Homologous genetic recombination: the pieces begin to fall into place. Crit Rev Microbiol 20: 125–142 [DOI] [PubMed] [Google Scholar]
- Courcelle J, Hanawalt PC (2003) RecA-dependent recovery of arrested DNA replication forks. Annu Rev Genet 37: 611–646 [DOI] [PubMed] [Google Scholar]
- Dutreix M, Moreau PL, Bailone A, Galibert F, Battista JR, Walker GC, Devoret R (1989) New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J Bacteriol 171: 2415–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehn M, Nilsson P, Uhlen M, Hober S (2001) Overexpression, rapid isolation, and biochemical characterization of Escherichia coli single-stranded DNA-binding protein. Protein Expr Purif 22: 120–127 [DOI] [PubMed] [Google Scholar]
- Frank EG, Gonzalez M, Ennis DG, Levine AS, Woodgate R (1996) In vivo stability of the Umu mutagenesis proteins: a major role for RecA. J Bacteriol 178: 3550–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Lehmann AR, Fuchs RP (2005) Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell 18: 499–505 [DOI] [PubMed] [Google Scholar]
- Fuchs RP, Fujii S, Wagner J (2004) Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem 69: 229–264 [DOI] [PubMed] [Google Scholar]
- Fujii S, Fuchs RP (2004) Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J 23: 4342–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Gasser V, Fuchs RP (2004) The biochemical requirements of DNA polymerase V-mediated translesion synthesis revisited. J Mol Biol 341: 405–417 [DOI] [PubMed] [Google Scholar]
- Gasior SL, Olivares H, Ear U, Hari DM, Weichselbaum R, Bishop DK (2001) Assembly of RecA-like recombinases: distinct roles for mediator proteins in mitosis and meiosis. Proc Natl Acad Sci USA 98: 8411–8418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem 71: 17–50 [DOI] [PubMed] [Google Scholar]
- Griffin TJT, Kolodner RD (1990) Purification and preliminary characterization of the Escherichia coli K-12 recF protein. J Bacteriol 172: 6291–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S, Sandler SJ, Clark AJ, Madiraju MV (1995) recO and recR mutations delay induction of the SOS response in Escherichia coli. Mol Gen Genet 246: 254–258 [DOI] [PubMed] [Google Scholar]
- Heller RC, Marians KJ (2006) Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439: 557–562 [DOI] [PubMed] [Google Scholar]
- Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H (2003) Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 8: 437–449 [DOI] [PubMed] [Google Scholar]
- Joo C, McKinney SA, Nakamura M, Rasnik I, Myong S, Ha T (2006) Real-time observation of RecA filament dynamics with single monomer resolution. Cell 126: 515–527 [DOI] [PubMed] [Google Scholar]
- Karu AE, Belk ED (1982) Induction of E. coli recA protein via recBC and alternate pathways: quantitation by enzyme-linked immunosorbent assay (ELISA). Mol Gen Genet 185: 275–282 [DOI] [PubMed] [Google Scholar]
- Kato T, Rothman RH, Clark AJ (1977) Analysis of the role of recombination and repair in mutagenesis of Escherichia coli by UV irradiation. Genetics 87: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC, Clow J, Somani R, Varghese A (1987) Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single-stranded DNA. Demonstration of competitive binding and the lack of a specific protein–protein interaction. J Mol Biol 193: 81–95 [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC, Krupp RA (1987) Effects of Escherichia coli SSB protein on the single-stranded DNA-dependent ATPase activity of Escherichia coli RecA protein. Evidence that SSB protein facilitates the binding of RecA protein to regions of secondary structure within single-stranded DNA. J Mol Biol 193: 97–113 [DOI] [PubMed] [Google Scholar]
- Kuzminov A (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63: 751–813, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston LD, O'Donnell M (2006) DNA replication: keep moving and don't mind the gap. Mol Cell 23: 155–160 [DOI] [PubMed] [Google Scholar]
- Lavery PE, Kowalczykowski SC (1992) Biochemical basis of the constitutive repressor cleavage activity of recA730 protein. A comparison to recA441 and recA803 proteins. J Biol Chem 267: 20648–20658 [PubMed] [Google Scholar]
- Lehmann AR, Fuchs RP (2006) Gaps and forks in DNA replication: rediscovering old models. DNA Repair (in press) [DOI] [PubMed] [Google Scholar]
- Liu YH, Cheng AJ, Wang TC (1998) Involvement of recF, recO, and recR genes in UV-radiation mutagenesis of Escherichia coli. J Bacteriol 180: 1766–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- Luisi-DeLuca C, Kolodner R (1994) Purification and characterization of the Escherichia coli RecO protein. Renaturation of complementary single-stranded DNA molecules catalyzed by the RecO protein. J Mol Biol 236: 124–138 [DOI] [PubMed] [Google Scholar]
- Lusetti SL, Hobbs MD, Stohl EA, Chitteni-Pattu S, Inman RB, Seifert HS, Cox MM (2006) The RecF protein antagonizes RecX function via direct interaction. Mol Cell 21: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju MV, Templin A, Clark AJ (1988) Properties of a mutant recA-encoded protein reveal a possible role for Escherichia coli recF-encoded protein in genetic recombination. Proc Natl Acad Sci USA 85: 6592–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi AA, Lloyd RG (1989) Identification of the recR locus of Escherichia coli K-12 and analysis of its role in recombination and DNA repair. Mol Gen Genet 216: 503–510 [DOI] [PubMed] [Google Scholar]
- McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC (2005) Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20: 783–792 [DOI] [PubMed] [Google Scholar]
- McInerney P, O'Donnell M (2004) Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J Biol Chem 279: 21543–21551 [DOI] [PubMed] [Google Scholar]
- Morimatsu K, Kowalczykowski SC (2003) RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell 11: 1337–1347 [DOI] [PubMed] [Google Scholar]
- Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP (2000) All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J 19: 6259–6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages V, Fuchs RP (2003) Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 300: 1300–1303 [DOI] [PubMed] [Google Scholar]
- Pham P, Bertram JG, O'Donnell M, Woodgate R, Goodman MF (2001) A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 409: 366–370 [DOI] [PubMed] [Google Scholar]
- Register JC III, Griffith J (1985) The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J Biol Chem 260: 12308–12312 [PubMed] [Google Scholar]
- Reuven NB, Arad G, Stasiak AZ, Stasiak A, Livneh Z (2001) Lesion bypass by the Escherichia coli DNA polymerase V requires assembly of a RecA nucleoprotein filament. J Biol Chem 276: 5511–5517 [DOI] [PubMed] [Google Scholar]
- Rupp WD, Howard-Flanders P (1968) Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol 31: 291–304 [DOI] [PubMed] [Google Scholar]
- Sandler SJ, Clark AJ (1994) RecOR suppression of recF mutant phenotypes in Escherichia coli K-12. J Bacteriol 176: 3661–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawitzke JA, Stahl FW (1992) Phage lambda has an analog of Escherichia coli recO, recR and recF genes. Genetics 130: 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper RM, Glickman BW, Loeb LA (1982) Mutagenesis resulting from depurination is an SOS process. Mutat Res 106: 1–9 [DOI] [PubMed] [Google Scholar]
- Schlacher K, Cox MM, Woodgate R, Goodman MF (2006) RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature 442: 883–887 [DOI] [PubMed] [Google Scholar]
- Schlacher K, Leslie K, Wyman C, Woodgate R, Cox MM, Goodman MF (2005) DNA polymerase V and RecA protein, a minimal mutasome. Mol Cell 17: 561–572 [DOI] [PubMed] [Google Scholar]
- Shan Q, Bork JM, Webb BL, Inman RB, Cox MM (1997) RecA protein filaments: end-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol 265: 519–540 [DOI] [PubMed] [Google Scholar]
- Steinborn G (1978) Uvm mutants of E. coli K12 deficient in UV mutagenesis. 1. Isolation of uvmmutants and their phenotypical characterization in DNA repair and mutagenesis. Mol Gen Genet 165: 87–93 [DOI] [PubMed] [Google Scholar]
- Sweasy JB, Witkin EM, Sinha N, Roegner-Maniscalco V (1990) RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J Bacteriol 172: 3030–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu K, Chi NW, Kolodner RD (1993) Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci USA 90: 3875–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu K, Kolodner RD (1994) Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem 269: 30005–30013 [PubMed] [Google Scholar]
- Wang TC, Chang HY, Hung JL (1993) Cosuppression of recF, recR and recO mutations by mutant recA alleles in Escherichia coli cells. Mutat Res 294: 157–166 [DOI] [PubMed] [Google Scholar]
- Whitby MC, Lloyd RG (1995) Altered SOS induction associated with mutations in recF, recO and recR. Mol Gen Genet 246: 174–179 [DOI] [PubMed] [Google Scholar]
- Williams KR, Murphy JB, Chase JW (1984) Characterization of the structural and functional defect in the Escherichia coli single-stranded DNA binding protein encoded by the ssb-1 mutant gene. Expression of the ssb-1 gene under lambda pL regulation. J Biol Chem 259: 11804–11811 [PubMed] [Google Scholar]
- Wood RD, Stein J (1986) Role of the RecF gene product in UV mutagenesis of lambda phage. Mol Gen Genet 204: 82–84 [DOI] [PubMed] [Google Scholar]
- Xu L, Marians KJ (2003) PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11: 817–826 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Figures S1, S2, S4 and S5