

Abstract
The redox siblings nitroxyl (HNO) and nitric oxide (NO) have often been
assumed to undergo casual redox reactions in biological systems. However,
several recent studies have demonstrated distinct pharmacological effects for
donors of these two species. Here, infusion of the HNO donor Angeli's salt
into normal dogs resulted in elevated plasma levels of calcitonin gene-related
peptide, whereas neither the NO donor diethylamine/NONOate nor the
nitrovasodilator nitroglycerin had an appreciable effect on basal levels.
Conversely, plasma cGMP was increased by infusion of diethylamine/NONOate or
nitroglycerin but was unaffected by Angeli's salt. These results suggest the
existence of two mutually exclusive response pathways that involve stimulated
release of discrete signaling agents from HNO and NO. In light of both the
observed dichotomy of HNO and NO and the recent determination that, in
contrast to the O2/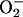 couple, HNO is a weak reductant, the relative reactivity of HNO with common
biomolecules was determined. This analysis suggests that under biological
conditions, the lifetime of HNO with respect to oxidation to NO, dimerization,
or reaction with O2 is much longer than previously assumed. Rather,
HNO is predicted to principally undergo addition reactions with thiols and
ferric proteins. Calcitonin gene-related peptide release is suggested to occur
via altered calcium channel function through binding of HNO to a ferric or
thiol site. The orthogonality of HNO and NO may be due to differential
reactivity toward metals and thiols and in the cardiovascular system, may
ultimately be driven by respective alteration of cAMP and cGMP levels.
couple, HNO is a weak reductant, the relative reactivity of HNO with common
biomolecules was determined. This analysis suggests that under biological
conditions, the lifetime of HNO with respect to oxidation to NO, dimerization,
or reaction with O2 is much longer than previously assumed. Rather,
HNO is predicted to principally undergo addition reactions with thiols and
ferric proteins. Calcitonin gene-related peptide release is suggested to occur
via altered calcium channel function through binding of HNO to a ferric or
thiol site. The orthogonality of HNO and NO may be due to differential
reactivity toward metals and thiols and in the cardiovascular system, may
ultimately be driven by respective alteration of cAMP and cGMP levels.
Keywords: Angeli's salt, superoxide dismutase, heme protein, cGMP, calcitonin gene-related peptide
During the last two decades, discussion of the chemistry of nitric oxide
(NO) in biological systems has primarily focused on the nitrosylation of heme
proteins such as soluble guanylyl cyclase and the production of reactive
nitrogen oxide species (RNOS)
(1–3).
The RNOS literature has largely been concerned with nitrogen dioxide
(NO2), dinitrogen trioxide (N2O3), and
peroxynitrite (ONOO–), which are formed through reaction with
molecular oxygen or superoxide (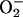 )
(4–6).
Recently, however, there has been increased interest in the one-electron
reduction product of NO, nitroxyl (HNO/NO–; nitrosyl
hydride/nitroxyl anion). Of particular note are studies suggesting that
oxidation of l-arginine by NO synthase (NOS) leads to production of
nitroxyl rather than NO under certain conditions
(7–10).
In this light, elucidation of the chemical biology of nitroxyl has acquired
new importance.
)
(4–6).
Recently, however, there has been increased interest in the one-electron
reduction product of NO, nitroxyl (HNO/NO–; nitrosyl
hydride/nitroxyl anion). Of particular note are studies suggesting that
oxidation of l-arginine by NO synthase (NOS) leads to production of
nitroxyl rather than NO under certain conditions
(7–10).
In this light, elucidation of the chemical biology of nitroxyl has acquired
new importance.
Comparisons of the toxicological and pharmacological properties of nitrogen oxide donor compounds have revealed that NO and HNO in general elicit distinct responses under a variety of biological conditions. In vitro, HNO reacts with O2 to generate potent oxidizing species capable of cleaving DNA, thereby augmenting oxidative damage (3, 11). The RNOS formed by NO autoxidation do not cause these cellular alterations under similar conditions (3, 12, 13). Rather, NO is an antioxidant, scavenging the reactive intermediates mediating oxidative stress. HNO has also been found to be a thiophilic electrophile (14–16), readily capable of modifying cellular thiol functions (14, 15). Conversely, NO reacts only indirectly with thiols after RNOS formation (17).
Contrasting effects are also apparent in vivo or ex vivo, for example in models of ischemia reperfusion injury. Exposure to NO donors at the onset of reperfusion provides protection against reperfusion injury in the heart and other organs (18–20). In contrast, HNO donors dramatically increase the infarct area and tissue injury in this methodology (21). Interestingly, exposure of the heart to HNO donors before ischemia resulted in protection against injury (22). The preconditioning effects were considerably lower with NO donors. These and other examples suggest that the physiological properties of NO and HNO are orthogonal (i.e., of the same origin but nonoverlapping), and that the biological response to either species is highly condition dependent.
Although redox conversion between HNO and NO can occur in vitro (7, 9, 23–26), the differential pharmacological effects of nitrogen oxide donors suggest both that interchange between HNO and NO is not facile in vivo and that each species interacts with distinct cellular targets. Here, we have examined the relative reactivity of HNO with common biomolecules in an attempt to elucidate the chemical basis for the orthogonal properties of NO and HNO in vivo, particularly in the cardiovascular system.
Methods
Angeli's salt (AS) and the diethylamine/NO adduct (DEA/NO) were synthesized and used as described (3, 13). Unless otherwise noted, chemicals were purchased from Sigma–Aldrich and were used without further purification. The assay buffer contained the metal chelator diethylenetriaminepentaacetic acid (DTPA, 50 μM) in calcium and magnesium-free Dulbecco's PBS (pH 7.4; GIBCO/BRL). All reactions were performed at 37°C. Additional experimental details are published as supporting information on the PNAS web site, www.pnas.org.
Plasma Analysis. The protocol was performed by using adult male dogs and was approved by the Animal Care and Use Committee of The Johns Hopkins University (27, 28). Arterial, venous, and coronary sinus plasma cGMP was detected by enzyme immunoassay (Biotrak, Amersham Pharmacia), and calcitonin gene-related peptide (CGRP) was assessed by radio immunoassay (Peninsula Laboratories) by using CGRP antiserum (RAS 6012).
Heme Protein Assays. Oxidation by AS of the ferric heme proteins ferricytochrome c (ferricyt c; horse heart), ferric myoglobin (metMb; horse heart) and horseradish peroxidase (HRP) was performed in deaerated assay buffer. The rate of oxidation by AS was monitored at 550 nm for ferricyt c, 575 nm for metMb, and 426 nm for HRP. The change in absorbance was determined from the amplitude of the exponential fit for each data set. Reduction by AS of oxymyoglobin [oxyMb; prepared as described (29)] was performed aerobically. The data were compared as ΔAbs at 426 or 582 nm for paired triplicate sets with and without AS (1-h exposure).
Results
Recently, HNO was found to mediate release of CGRP in the canine coronary system (27). Because it is often assumed that HNO and NO are readily interconverted in vivo, we sought to address whether HNO or NO was responsible for CGRP release in the cardiovascular system. To this end, the levels of CGRP in canine plasma were measured after 10-min treatment with HNO and NO donors. Administration of AS resulted in elevated CGRP levels, whereas DEA/NO and the nitrovasodilator nitroglycerin (NTG; American Reagent Laboratories, Shirley, NY) did not significantly increase CGRP over baseline (Table 1). This suggests that HNO alone is responsible for CGRP release and that HNO is not an intermediate in NTG metabolism.
Table 1. Comparison of plasma levels of CGRP and cGMP elicited by 10-min infusion of NO and HNO donors.
|
Artery
|
Vein
|
||||||
|---|---|---|---|---|---|---|---|
| Donor | n | Infusion rate, μg/kg/min | Baseline | Post infusion | Baseline | Post infusion | |
| CGRP, pg/ml | AS | 7 | 10 | 25.8 ± 1.5 | 52.3 ± 4.7* | 27.8 ± 1.3 | 42.5 ± 1.3* |
| DEA/NO | 3 | 2 | 28.1 ± 3.2 | 30.6 ± 5.2 | 21.5 ± 1.5 | 22.0 ± 1.0 | |
| NTG | 4 | 10 | 29.4 ± 4.9 | 28.9 ± 2.6 | 26.3 ± 5.0 | 23.1 ± 6.1 | |
| total | 42 | 28.5 ± 1.1 | 28.3 ± 1.2 | ||||
| cGMP, nM | AS | 6 | 10 | 22.5 ± 8.5 | 20.2 ± 5.7 | 20.3 ± 8.1 | 23.2 ± 8.2 |
| DEA/NO | 1 | 3.1 | 16.1 | 66.9 | 26.7 | 65.4 | |
| NTG | 1 | 3.3 | 29.7 | 49.3 | 27.3 | 45.4 | |
| NTG | 1 | 10 | 30.6 | 52.6 | 28.5 | 52.5 | |
| NTG | 1 | 16 | 29.7 | 63.0 | 27.3 | 70.1 | |
P < 0.01 vs. baseline. Coronary sinus data are provided in the supporting information.
Conversely, plasma levels of cGMP were not altered by AS but were elevated by comparable amounts of DEA/NO and NTG. In a similar model, NTG (20 μg/kg per min) was previously shown to stimulate cGMP formation [from 42.7 to 110 nM in coronary artery plasma (30)]. These observations strongly suggest that HNO and NO participate in distinct cardiovascular pathways through respective activation of CGRP and cGMP. Thus, measurement of elevated plasma levels of CGRP may provide a unique signature for HNO-mediated signaling in the vascular system.
The chemical biology of HNO (13) is gradually being elucidated. The direct reactions of HNO under biological conditions are significantly more varied than for NO (4, 31) and result in both oxidation and reduction of biomolecules. In analogy to NO (31), reaction with O2 results in RNOS formation. In the absence of other reaction partners, HNO can also dimerize to produce N2O after dehydration (32). The biochemical/pharmacological outcome of HNO production will thus depend on the balance achieved between scavenging and activating/deactivating pathways. The orthogonality of HNO and NO donors in vivo as well as the distinctive elevation of CGRP or cGMP suggests that reactions with biomolecules that are capable of interconverting HNO and NO in vitro such as Cu,Zn superoxide dismutase (CuZnSOD) and cytochrome c are kinetically constrained in vivo, as has been suggested (33).
To identify reactions that may account for specific HNO-mediated CGRP release and specific stimulation of cGMP production by nitrovasodilators, the relative reactivity of HNO with key biomolecules was assessed. The relative reactivity of various compounds for a specific species can be obtained from comparison of the concentrations of reactants ([S]0.5) required to achieve 50% of the maximal rate of product formation under controlled conditions. Similarly, direct in situ comparisons can be made from competition experiments when the quenching reactant ([Q]0.5) does not interfere with detection of the product. Under these conditions, the rate of monitored product formation is equivalent to the rate(s) of consumption leading to nonobserved product(s) (34). Generally, linear double reciprocal plots of product vs. reactant concentrations or linear single reciprocal plots of product vs. quencher concentrations can provide these values. Unfortunately, HNO is involved in numerous side reactions, and these reciprocal plots do not always have linear solutions. However, a more qualitative description of the relative rates can be obtained by approximating the concentration of reactant or quencher at 50% maximal product.
In general, significant reactivity would be expected between HNO and any ferric heme protein, leading to reductive nitrosylation of the heme (29)
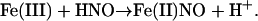 |
[1] |
Typically, ferrous–nitrosyl complexes are rather stable; however, the unusually low stability of the NO-heme adduct in cytochrome c (cyt c) results in release of NO, thus catalyzing the oxidation of HNO to NO (14, 25). In the presence of cyt c under anaerobic conditions, the competing reaction (HNO consumption) is presumed to occur only through dimerization, and thus the [S]0.5 is representative of the relative rates of ferricyt c reduction and HNO dimerization (Scheme 2 in supporting information).
Product formation as a result of exposure of AS (5 μM) to varied ferricyt c (2–100 μM) is shown in Fig. 1A. The point of 50% product formation was estimated to correspond to half the amplitude of the change in ΔAbs obtained from an exponential fit of the data including the origin. In this fashion, [S]0.5, from the x-axis value at 50% product, is estimated to be 4 ± 0.5 μM ferricyt c. This value should be considered to be only a rough approximation, in part because it falls below the usable concentration range of ferricyt c.
Fig. 1.

Oxidation of ferricyt c by AS. (A) Effect of constant AS (5 μM final) and varied ferricyt c (2–100 μM). (B) Effect of varied AS (5–200 μM) and constant ferricyt c (100 μM).
When the conditions are reversed with constant ferricyt c (100 μM) and varied AS (5–200 μM), the analysis must be altered. In this case, the equivalence point occurs when the product concentrations of the two pathways are equivalent. Because the assumption is made that no other pathways contribute to HNO consumption, the sum of the product concentrations ([ferrocyt c] + 2[N2O]) equals the initial concentration of AS. Thus, the equivalence point is located where the measured product equals one-half the initial concentration of AS. This value ([AS]0.5) is estimated from Fig. 1B to be 50 ± 5 μM AS (resulting in 25 μM ferrocyt c production).
Liochev and Fridovich (25) determined the relative rates of the reactions of ferricyt c and CuZnSOD with HNO via a competitive kinetic analysis at 23°C. Similarly, we examined the relative quenching of the reduction of ferricyt c (100 μM) by AS (25 μM) by different biomolecules at 37°C.
Increasing CuZnSOD (1–100 μM) reduced the yield of ferrocyt c (data not shown). Interestingly, the reaction of ferricyt c with HNO in the presence of CuZnSOD appeared to be catalytic (mechanism discussed in supporting information) such that ferrocyt c formation plateaued at approximately one-third the value obtained in the absence of CuZnSOD. The catalytic reoxidation of CuZnSOD hampers determination of [Q]0.5. However, the preplateau ΔAbs values approached 50% of the original ΔAbs, allowing an estimation of [Q]0.5 at 4 ± 1 μM CuZnSOD (Table 2). The ratio of the concentrations of ferricyt c to SOD at 50% product, [S]/[Q], is therefore ≈25. Assuming that other consumptive pathways, such as HNO dimerization, are negligible in competition assays of this type given the relatively low concentration of AS used, this ratio corresponds to the ratio of the rate constants for reaction of HNO with SOD or ferricyt c. In general terms, when the reactant (S) is in large excess of the quenching scavenger (Q), the relationship is
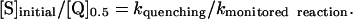 |
[2] |
Similar catalytic behavior was observed with the SOD mimic Tempol (0.05–5 mM). Quenching by MnSOD (1–45 μM), however, was well behaved with 6 ± 1 μM MnSOD required to quench ferricyt c reduction by 50% (data not shown).
Table 2. Concentration of scavenger required for ≈50% quenching and relative ratio of reactant concentrations.
| Reactant (S) | Scavenger (Q) | 50% quenching, μM | Relative ratio ([S]/[Q]) |
|---|---|---|---|
| Ferricyt c (100 μM) | CuZnSOD | 4 ± 1* | 25 |
| MnSOD | 6 ± 1 | 17 | |
| GSH | <4* | >25 | |
| metMb (50 μM) | CuZnSOD | 60 | 0.8 |
| GSH | 20 | 2.5 | |
| Tempol | 500 | 0.1 | |
| HRP (50 μM) | GSH | 50 | 1.0 |
| oxyMb (40 μM) | GSH | 200 ± 30 | 0.2 |
| NAC | 850 ± 75 | 0.05 | |
| O2 (180 μM) | CuZnSOD | 0.8 ± 0.2 | 225 |
| GSH | ∼0.3* | 600 | |
| NAC | 1 ± 0.2 | 180 | |
| catalase | 2 ± 0.4 | 90 |
Approximate values due to constraints of the experimental methods.
The complications arising from release of NO from ferrocyt c would not be expected from other ferric hemes. Myoglobin (Mb) was included in the analysis because it contains a single heme and, with the exception of human Mb, no thiols, thus avoiding the complexity of HNO chemistry that would be expected with hemoglobin. Additionally, both the ferric and oxy states of Mb react with HNO (Eqs. 1 and 3), providing an interesting comparative system.
The relative scavenging abilities of CuZnSOD (20–100 μM) and Tempol (0.25–2 mM) for reductive nitrosylation of metMb (50 μM) by AS (25 μM) are reported in Table 2. The ratio of metMb to SOD concentration at 50% quenching is approximately equimolar, indicating that the rate constants are of similar magnitude. The [S]/[Q] ratio was 10-fold higher for Tempol, implying that this SOD mimetic is not as reactive toward HNO as the protein.
Quenching by reduced glutathione (GSH) was also examined, because thiols are known to react readily with HNO (15). Addition of stoichiometric amounts of GSH and AS (25 μM) to ferricyt c (100 μM) resulted in >90% quenching of heme reduction compared with AS alone (data not shown), indicating that the rate of reaction of HNO with GSH is considerably faster than with ferricyt c (≫5-fold). Experimental constraints did not permit accurate determination of [Q]0.5 but did allow for estimation of a maximum value of 4 μM GSH (>25-fold difference in rate; Table 2).
The concentration ratio for GSH to ferric heme protein was only 2.5 for metMb and 1.0 for HRP (Table 2; 50 μM metMb or HRP, 25 μM AS, 10–100 μM GSH), implying that reactions of HNO with GSH, metMb and HRP all have similar rate constants. Interestingly, the GSH (20–2000 μM) to oxyMb (40 μM) ratio was 0.2, suggesting that the reaction of HNO (20 μM AS) with oxyMb (14)
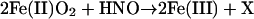 |
[3] |
is significantly faster than with metMb (Eq. 1).
Under aerobic conditions, decomposition of AS (25 μM) in the presence of ferricyt c (100 μM) did not produce reduced product, as previously demonstrated (25), suggesting that 200 μM O2 out-competes 100 μM ferricyt c for HNO. The product of the reaction between HNO and O2 has not been characterized (13); however, the effects of this reaction are quite distinct from those of HNO alone (3, 35). The HNO/O2 product has previously been shown to oxidize dihydrorhodamine 123 (DHR; Molecular Probes) by two electrons to form the fluorescent dye rhodamine 123 (RH) (13). This fluorescent assay (described in detail in ref. 13) has the advantage of allowing considerably lower, more pharmacologically/physiologically relevant concentrations of AS to be used compared with monitoring changes in heme absorbance.
Because the oxidant formed from the HNO/O2 reaction has a high affinity for DHR (13), under the experimental conditions >95% of the HNO/O2 product formed will react with DHR. Thus, the relative ratio of reaction of HNO with O2 vs. dimerization could be determined by varying AS (0.5–50 μM) in the presence of excess DHR (25 μM). From the plot of RH fluorescence vs. AS concentration (Fig. 2A), [AS]0.5 is estimated to be 11 ± 1 μM AS. From Fig. 2A, it can be concluded that under normoxic conditions, dimerization will be the predominant pathway above 30 μM AS (plateau point), whereas below 5 μM AS (linear portion of curve), >99% of HNO will be consumed by O2. Thus, the assumption that HNO dimerization will be negligible in the protein competition experiments described above appears to be valid.
Fig. 2.

Oxidation of DHR by AS. (A) Varied amounts of AS (0.5–50 μM final) were added to assay buffer containing DHR (25 μM). (B) Aliquots of AS (0.5 μM final) were added to assay buffer containing DHR (25 μM) and varying amounts of GSH, NAC, CuZnSOD (all 0.5–8 μM), or catalase (0.5-5 μM).
In the presence of 25 μM DHR and 0.5 μM AS, quenching of AS-mediated DHR oxidation by thiols (0.5–8 μM) and metalloproteins was concentration-dependent. Under these conditions, dimerization is excluded as a kinetically viable pathway, which results in a linear solution to reciprocal plots of RH fluorescence to quencher concentration (Fig. 2B; mathematical derivation in ref. 34). The [Q]0.5 values can thus be determined with a higher degree of confidence than for the heme protein experiments, from the negative X-intercept values (Table 2).
Ferricyt c, metMb, and HRP were not included in this analysis due to complications arising from increased fluorescence in control solutions. Similarly, at >10 μM catalase, significant oxidation of DHR was observed in the absence of AS. However, a [Q]0.5 value of 2 μM was obtained with 0.5–5 μM catalase (Fig. 2B). Quenching by CuZnSOD (0.5–8 μM) reached a plateau at ≈80–90%, suggesting that a product formed from the reaction of CuZnSOD and HNO also oxidizes DHR but with considerably less efficiency than the HNO/O2 product. Because aerobic production of NO does result in RH formation (13), the conclusion was made that the residual RH signal is due to the presence of NO produced by CuZnSOD oxidation of HNO. Subtraction of the estimated DHR concentration at infinite CuZnSOD (by exponential fit) gave a linear reciprocal plot with a [Q]0.5 value of 0.8 ± 0.2 μM CuZnSOD (Fig. 2B). The >3-fold difference between [Q]0.5 for GSH (0.3 μM) and N-acetylcysteine (NAC; 1 μM) is consistent with the results found in quenching of oxyMb oxidation above (Table 2).
Kinetic Evaluation. Table 2 provides a comparison of the affinity of HNO for different biomolecules. From these data, the relative reactivity toward HNO is oxyMb > GSH, HRP > NAC, CuZnSOD, MnSOD, metMb ≈ catalase > Tempol, ferricyt c > O2. To quantify the rate constants, a known value is required. Dimerization of HNO has been estimated to have a rate constant of 4 × 109 M–1·s–1 (32), based on a pKa of 4.7 (36). Recently, however, Shafirovich and Lymar (37) reported a revised rate constant of 8 × 106 M–1·s–1 using flash photolysis techniques. If we assume that the data in Fig. 1 are a function only of competition between reduction of ferricyt c and dimerization (Scheme 2 in supporting information), at the point of 50% product formation, the rates of each pathway are identical
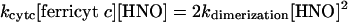 |
[4] |
or more generally,
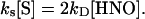 |
[5] |
To solve for kS, [HNO] must be known. Assuming a steady state of HNO and a quadratic solution (see supporting information for complete derivation and discussion of the kinetic evaluation),
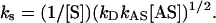 |
[6] |
From Fig. 1 A and B, kS for ferricyt c is estimated to be 4 × 104 M–1·s–1. Additionally, the rate constant for the reaction between O2 and HNO is calculated from Fig. 2A to be 3 × 103 M–1·s–1. Because this derived value is an order of magnitude lower that that for ferricyt c, the complete quenching of HNO-mediated ferricyt c reduction observed in aerobic buffer is likely not strictly a function of kinetics, and secondary reactions may be significant.
From the rate constants derived for ferricyt c and O2 and the ratio of concentrations given in Table 2, the rates constants (Table 3) for the other biomolecules can now be calculated from Eq. 2. Certainly, these values should be considered to be only predictive, with a degree of uncertainty that will be refined with direct measurements using rapid time-resolved techniques. However, within the approximations made, these values offer insight into the reactions of HNO that are kinetically viable in biological systems.
Table 3. Derived rate constants.
|
k (M-1·s-1) determined from
|
||
|---|---|---|
| Biomolecule | Ferricyt c | O2 |
| Ferricyt c | 4 × 104 | |
| 2 × 104 [23°C (25)]* | ||
| O2 | 3 × 103 | |
| CuZnSOD | 1 × 106 | 7 × 105 |
| 9 × 104 [23°C (25)]* | ||
| MnSOD | 7 × 105 | |
| GSH | 2 × 106 | |
| NAC | 5 × 105 | |
| Catalase† | 3 × 105 | |
| metMb† | 8 × 105 | |
| oxyMb† | 1 × 107 | |
| HRP† | 2 × 106 | |
| Tempol‡ | 8 × 104 | |
Recalculated value based on a rate constant for dimerization of 8 × 106 M-1·s-1 (37).
Secondary derivation from O2 → GSH.
Tertiary derivation from O2 → GSH → metMb.
Discussion
The cardiovascular system plays a central role in maintenance of organ homeostasis, and the initially surprising discovery of the function of NO in regulation of vascular tone via stimulation of guanylyl cyclase (1, 2) ignited into a highly prolific area of biomedical research. Recently, nitroxyl, a potential intermediate in NOS biology (7–10), has been shown to regulate release of another vasoactive substance CGRP (27, 38). To date, comparative studies of the in vitro, in vivo, and ex vivo effects of NO and HNO donors have universally revealed orthogonal responses to these two nitrogen oxides (for example, refs. 3, 21, 27, 35, and 39). These results prompted comparison of CGRP and cGMP levels in canine plasma after infusion of NO and HNO donors. Although elevated levels of CGRP were observed only with the HNO donor AS (Table 1), plasma levels of cGMP were increased by DEA/NO and NTG and not by AS.
These observations strongly suggest that HNO and NO participate in distinct cardiovascular signaling pathways through respective stimulation of CGRP and cGMP formation/release. Because CGRP and related peptides are known to act, at least in part, through activation of the cAMP pathway (40–42), we propose that the orthogonality of response in the cardiovascular system to HNO and NO is ultimately a function of the orthogonality in response to increases in cGMP and cAMP. Although nitroxyl production by NOS in vivo remains speculative, this intriguing possibility may open new avenues of investigation into disease etiology via dysregulation of nitrogen oxide production under pathophysiological conditions. Further, there is clear pharmacological potential for specific regulation by NO and HNO.
The orthogonality of biological response to HNO and NO donors leads to fundamental questions about the chemistry of these two redox siblings, particularly because in situ experiments (23–26) have suggested that interconversion of HNO and NO is facile. We sought to address the issue of interconvertibility and determine the specific reactivity expected for HNO under biological conditions.
The pKa for nitroxyl was originally reported as 4.7 (36), indicating that NO– was the predominant species at pH 7.4. Analogous to O2, the lowest energy state for NO– is the triplet, suggesting that NO– will react with O2 to produce ONOO– (43). However, the aerobic chemistry derived from AS was ascertained to be substantially dissimilar from that of synthetic ONOO– (13, 44), revealing inconsistencies with the generally accepted chemical model for nitroxyl. Recently, the acid-base equilibria of nitroxyl have been reevaluated, and the pKa for deprotonation of HNO is suggested to exceed 11 (37, 45). Therefore, HNO is now implicated as not only a significant but likely the exclusive species present in the acid/base equilibrium of HNO/NO– in biological systems.
A favorable reduction potential for the NO/NO– couple was
derived using the original literature pKa value of 4.7
(36) and the assumption that
the acid-base equilibrium involved only singlet species. This calculated value
(+0.39 V; ref. 43) indicated
that NO could be rapidly reduced to NO– through casual redox
chemistry, in analogy to conversion of O2 to
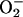 . However, the actual redox chemistry
is more complicated than that of a straightforward NO/NO–
couple due to the high pKa of HNO and the fact that the protonated
and unprotonated ground state species have different spin states
(1HNO and 3NO–). The reduction
potential of NO has recently been determined to be lower than –0.7 V vs.
normal hydrogen electrode (37,
45). This potential lies at
the high end of the biological redox scale for eukaryotic cells, indicating
nonfacility for outer-sphere electron transfer. Outer-sphere reactions result
from collision of two molecules, with the electron in essence jumping from the
oxidant to the reductant. The major factors that govern the efficiency of
these reactions are electron potential and distance. The high activation
barrier renders NO virtually inert to reduction with respect to physiological
events that readily reduce O2.
. However, the actual redox chemistry
is more complicated than that of a straightforward NO/NO–
couple due to the high pKa of HNO and the fact that the protonated
and unprotonated ground state species have different spin states
(1HNO and 3NO–). The reduction
potential of NO has recently been determined to be lower than –0.7 V vs.
normal hydrogen electrode (37,
45). This potential lies at
the high end of the biological redox scale for eukaryotic cells, indicating
nonfacility for outer-sphere electron transfer. Outer-sphere reactions result
from collision of two molecules, with the electron in essence jumping from the
oxidant to the reductant. The major factors that govern the efficiency of
these reactions are electron potential and distance. The high activation
barrier renders NO virtually inert to reduction with respect to physiological
events that readily reduce O2.
The reverse reaction, oxidation of NO– to NO, should, however, be facile, and anaerobic exposure of the nitroxyl donor methanesulfohydroxamic acid to a series of viologen acceptors indicated that the oxidation potential for NO– is close to 0.7 V (45). Alkyl and aryl sulfohydroxamic acids are relatively stable at physiological pH but rapidly liberate nitroxyl under basic conditions (36, 37), and the viologen studies were performed at pH >12. On the other hand, the decomposition rate of AS is significantly enhanced below pH 8 (46). Similar studies with AS but at pH 7 did not result in observable viologen reduction, indicating an effective potential of <0.4 V (45). This difference in reactivity suggests that 1HNO is the predominant species generated from AS at pH 7, whereas 3NO– is produced by methanesulfohydroxamic acid at pH 12. On this basis, we propose that at physiological pH the reactivity of nitroxyl originates from HNO regardless of the production source and that the chemistry of NO– will be observed only at high pH. Further, conversion of HNO to NO is established to be considerably less facile than oxidation of NO– to NO. The distinct reactivity profiles toward diaminofluoroscein observed within cells following exposure to AS or DEA/NO are consistent with these findings (39), as are the distinct bioactivity profiles of AS and Piloty's acid, which is a NO donor under physiological conditions (47).
Clearly, decomposition of AS does result in reduction chemistry as illustrated with ferricyt c and SOD (ref. 25; Fig. 1). The question thus arises whether HNO is oxidized to NO directly or through an equilibrium with NO–
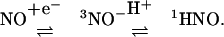 |
[7] |
It could be argued that even with a pKa > 11, reactions mediated through NO– may be kinetically viable. NO– chemistry from AS would depend on HNO deprotonation
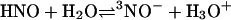 |
[8] |
In a preequilibrium solution, the pKa limits this reaction, effectively reducing even diffusion-controlled reactions by 104 at neutral pH (48). Due to the necessity of a spin flip, deprotonation of HNO to 3NO– has been shown by Shafirovich and Lymar (37) to be relatively slow proceeding with a rate constant of 5 × 104 M–1·s–1. This suggests that at neutral pH the half-life of 1 μM HNO with respect to deprotonation is >10 s. Since consumptive pathways such as dimerization and reactions with O2 or biomolecules are considerably faster than deprotonation, NO– would be expected to have little or no role in the biological chemistry of HNO.
Reduction of ferricyt c by AS (k ≈ 104
M–1·s–1) is
approximately two orders of magnitude slower than by
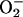 [k = 4 ×
106
M–1·s–1;
(49)]. Assuming these
reactions proceed through outer-sphere electron transfer, this suggests that
the HNO oxidation potential is considerably less than that of
[k = 4 ×
106
M–1·s–1;
(49)]. Assuming these
reactions proceed through outer-sphere electron transfer, this suggests that
the HNO oxidation potential is considerably less than that of
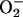 (+0.33 V vs. normal hydrogen
electrode). It can be argued that E° may be higher as the effective
potential will be limited by other reactions such as dimerization and reaction
with O2. However, these results suggest that HNO is at best a mild
reducing agent and therefore, it is unlikely that HNO is directly converted to
NO through simple outer-sphere electron transfer under biological
conditions.
(+0.33 V vs. normal hydrogen
electrode). It can be argued that E° may be higher as the effective
potential will be limited by other reactions such as dimerization and reaction
with O2. However, these results suggest that HNO is at best a mild
reducing agent and therefore, it is unlikely that HNO is directly converted to
NO through simple outer-sphere electron transfer under biological
conditions.
The potential for reduction of Cu(II) to Cu(I) in SOD is 0.4 V (50). The above limitation of 0.33 V for oxidation of HNO, indicates that HNO does not reduce SOD by outer-sphere electron transfer. Rather, an inner-sphere mechanism is suggested. In contrast to outer-sphere mechanisms, inner-sphere electron transfer depends on formation of a covalent bond before electron transfer. Often symmetry rules in addition to redox potentials are factors in the facility of these reactions. In the case of SOD, HNO would be expected to first bind to Cu(II) forming a Cu(I)NO complex, analogously to reductive nitrosylation of metMb (14) (Eq. 1). The resulting d10 configuration for copper would result in rapid elimination of NO. Thus, conversion of HNO to NO is likely not an incidental reaction but requires catalysis (inner-sphere, addition reactions) by SOD and ferric heme proteins or their mimics (Scheme 1). Other metal-mediated mechanisms may also exist (33).
Scheme 1.

From the relative reactivity of HNO established here (Table 3), a biological scheme can be logically developed. Cells and tissues are heterogeneous mixtures containing varied concentrations of GSH and metals. The potential for HNO and NO reactivity can be accessed based on the redox milieu of the cellular microenvironment. In the cytosol, the SOD concentration is ≈10 μM (51, 52) while that of GSH is generally 1–10 mM (53). Since the rate constants are similar, HNO would be expected to react with GSH rather than be converted to NO by CuZnSOD. Therefore, although interconversion of HNO and NO does occur in situ, it can be argued that these reactions are likely to be of little relevance for most cellular compartments due to kinetic constraints, as previously suggested (33). The significance of the HNO/O2 reaction may be similarly limited. Thus, in the cytoplasm and other cellular compartments rich in GSH such as the mitochondrion, HNO would be expected to have little opportunity to react with other targets.
The concentration of GSH is substantially lower in cell membranes while the solubilities of NO and O2 are increased ≈10-fold compared to the cytosol (54). The neutral species HNO would also be expected to be membrane soluble. In this environment, the longer lifetime with respect to consumption by GSH and other scavengers such as ascorbate (13) may allow modification of a target such as a ferric heme or a protein thiol, which similarly to NAC, likely will react more slowly with HNO than does GSH. Secondary (indirect) modifications of biomolecules by the HNO/O2 product also may be kinetically viable in membranes or within hydrophobic regions of proteins, consistent with the pattern of diaminofluoroscein reactivity observed within cells during aerobic AS decomposition (39). Fig. 2A indicates that HNO consumption via dimerization is unlikely to be significant even at the increased concentrations expected in hydrophobic areas.
In blood vessels, the high reactivity with ferric or oxyheme proteins would also be expected to reduce the diffusibility of HNO, similarly to NO. However, i.v. infusion of HNO and NO donors does elicit pharmacological responses even at low micromolar concentrations (27). The reactivity of HNO with free or red blood cell-encapsulated hemoglobin remains to be ascertained.
Our analysis suggests that the reactivity of HNO will be controlled by multiple consumption pathways and that diffusion will be restricted to an even greater extent than for NO. Thus, modifications are most likely to occur if HNO is produced either adjacent to the target molecule or in sequestered regions of the cell.
Interestingly, release of CGRP is mediated by an influx of calcium through voltage-gated calcium channels in nonadrenergic/noncholinergic neurons (55). Subsequent migration of CGRP to receptor activity-modifying protein receptors in myocytes can activate protein kinase A through a cAMP mechanism (56). The chemical biology of HNO elucidated here suggests that HNO mediates CGRP release through interaction at the channel itself, perhaps through binding to a metal or thiol. A similar mechanism for calcium release via interaction of HNO with a metal has been proposed for modulation of N-methyl-d-aspartate receptors (35).
Conclusion
Here, we have further elucidated the chemical biology of HNO through examination of the reactivity of HNO with various biological targets. This evaluation suggests that the lifetime of HNO with respect to oxidation to NO, dimerization, and reaction with O2 is much longer than previously assumed. As with NO, location and concentration will ultimately determine the biological effects of HNO. The effective scavenging by heme proteins and GSH should provide sufficient protection against excess HNO production. However, when HNO is produced in close proximity to the target or in areas of reduced metal or GSH content, HNO may have a sufficient lifetime to allow mediation of a biological effect. The differential behavior of the redox siblings HNO and NO is to a great extent a function of their specific chemistry toward distinct molecular targets. For instance, NO will bind to non-O2-binding cytosolic ferrous proteins, whereas HNO will preferentially interact in membranes and areas of low scavenger content with proteins containing ferric centers or thiols. Thus, the focal orthogonal chemistry of HNO and NO appears to be an ideal system to orchestrate global modulation of various pathways under diverse conditions.
Supplementary Material
Acknowledgments
We are grateful to Prof. Peter C. Ford for insightful discussions. N.P. was supported by a Beginning Grant-in-Aid (0265435U, Mid-Atlantic Affiliate) from the American Heart Association.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AS, Angeli's salt (Na2N2O3); CGRP, calcitonin gene-related peptide; ferricyt c, ferricytochrome c; DEA/NO, diethylamine/NO adduct ([Et2NN(O)NO]Na); DHR, dihydrorhodamine 123; GSH, reduced glutathione; HRP, horseradish peroxidase; metMb, ferric myoglobin; NAC, N-acetylcysteine; NOS, NO synthase; NTG, nitroglycerin; oxyMb, oxymyoglobin; RH, rhodamine 123; RNOS, reactive nitrogen oxide species; SOD, superoxide dismutase; HNO, nitrosyl hydride.
References
- 1.Murad, F. (1994) Recent Prog. Hormone Res. 49, 239–248. [DOI] [PubMed] [Google Scholar]
- 2.Ignarro, L. J. (1989) Pharm. Res. 6, 651–659. [DOI] [PubMed] [Google Scholar]
- 3.Wink, D. A., Feelisch, M., Fukuto, J., Chistodoulou, D., Jourd'heuil, D., Grisham, M. B., Vodovotz, Y., Cook, J. A., Krishna, M., DeGraff, W. G., et al. (1998) Arch. Biochem. Biophys. 351, 66–74. [DOI] [PubMed] [Google Scholar]
- 4.Miranda, K., Espey, M. G., Jourd'heuil, D., Grisham, M. B., Fukuto, M., Feelisch, M. & Wink, D. A. (2000) in Nitric Oxide: Biology and Pathobiology, ed. Ignarro, L. (Academic, San Diego), pp. 41–55.
- 5.Espey, M. G., Miranda, K. M., Feelisch, M., Fukuto, J., Grisham, M. B., Vitek, M. & Wink, D. A. (2000) Ann. N.Y. Acad. Sci. 899, 209–221. [DOI] [PubMed] [Google Scholar]
- 6.Grisham, M. B., Jourd'Heuil, D. & Wink, D. A. (1999) Am. J. Physiol. 276, G315–G321. [DOI] [PubMed] [Google Scholar]
- 7.Hobbs, A. J., Fukuto, J. M. & Ignarro, L. J. (1994) Proc. Natl. Acad. Sci. USA 91, 10992–10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pufahl, R. A., Wishnok, J. S. & Marletta, M. A. (1995) Biochemistry 34, 1930–1941. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt, H., Hofmann, H., Schindler, U., Shutenko, Z. S., Cunningham, D. D. & Feelisch, M. (1996) Proc. Natl. Acad. Sci. USA 93, 14492–14497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adak, S., Wang, Q. & Stuehr, D. J. (2000) J. Biol. Chem. 275, 33554–33561. [DOI] [PubMed] [Google Scholar]
- 11.Ohshima, H., Gilibert, I. & Bianchini, F. (1999) Free Radical Biol. Med. 26, 1305–1313. [DOI] [PubMed] [Google Scholar]
- 12.Chazotte-Aubert, L., Oikawa, S., Gilibert, I., Bianchini, F., Kawanishi, S. & Ohshima, H. (1999) J. Biol. Chem. 274, 20909–20915. [DOI] [PubMed] [Google Scholar]
- 13.Miranda, K. M., Espey, M. G., Yamada, K., Krishna, M., Ludwick, N., Kim, S., Jourd'heuil, D., Grisham, M. B., Feelisch, M., Fukuto, J. M., et al. (2001) J. Biol. Chem. 276, 1720–1727. [DOI] [PubMed] [Google Scholar]
- 14.Doyle, M. P., Mahapatro, S. N., Broene, R. D. & Guy, J. K. (1988) J. Am. Chem. Soc. 110, 593–599. [Google Scholar]
- 15.Wong, P. S. Y., Hyun, J., Fukuto, J. M., Shirota, F. N., DeMaster, E. G., Shoeman, D. W. & Nagasawa, H. T. (1998) Biochemistry 37, 5362–5371. [DOI] [PubMed] [Google Scholar]
- 16.Bartberger, M. D., Fukuto, J. M. & Houk, K. N. (2001) Proc. Natl. Acad. Sci. USA 98, 2194–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wink, D. A., Nims, R. W., Darbyshire, J. F., Christodoulou, D., Hanbauer, I., Cox, G. W., Laval, F., Laval, J., Cook, J. A., Krishna, M. C., et al. (1994) Chem. Res. Toxicol. 7, 519–525. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, G., Tsao, P. S. & Lefer, A. M. (1991) Crit. Care Med. 19, 244–252. [DOI] [PubMed] [Google Scholar]
- 19.Kubes, P., Suzuki, M. & Granger, D. N. (1991) Proc. Natl. Acad. Sci. USA 88, 4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mason, R. B., Pluta, R. M., Walbridge, S., Wink, D. A., Oldfield, E. H. & Boock, R. J. (2000) J. Neurosurg. 93, 99–107. [DOI] [PubMed] [Google Scholar]
- 21.Ma, X. L., Cao, F., Liu, G. L., Lopez, B. L., Christopher, T. A., Fukuto, J. M., Wink, D. A. & Feelisch, M. (1999) Proc. Natl. Acad. Sci. USA 96, 14617–14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pagliaro, P., Mancardi, D., Rastaldo, R., Penna, C., Gattullo, D., Miranda, K. M., Feelisch, M., Wink, D. A., Kass, D. A. & Paolocci, N. (2003) Free Radical Biol. Med. 34, 33–43. [DOI] [PubMed] [Google Scholar]
- 23.Murphy, M. E. & Sies, H. (1991) Proc. Natl. Acad. Sci. USA 88, 10860–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuto, J. M., Hobbs, A. J. & Ignarro, L. J. (1993) Biochem. Biophys. Res. Commun. 196, 707–713. [DOI] [PubMed] [Google Scholar]
- 25.Liochev, S. I. & Fridovich, I. (2002) Arch. Biochem. Biophys. 402, 166–171. [DOI] [PubMed] [Google Scholar]
- 26.Sharpe, M. A. & Cooper, C. E. (1998) Biochem. J. 332, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paolocci, N., Saavedra, W. F., Miranda, K. M., Martignani, C., Isoda, T., Hare, J. M., Espey, M. G., Fukuto, J. M., Feelisch, M., Wink, D. A., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10463–10468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senzaki, H., Isoda, T., Paolocci, N., Ekelund, U., Hare, J. M. & Kass, D. A. (2002) Circulation 101, 1040–1048. [DOI] [PubMed] [Google Scholar]
- 29.Miranda, K. M., Nims, R. W., Thomas, D. D., Espey, M. G., Citrin, D., Bartberger, M. D., Paolocci, N., Fukuto, J. M., Feelisch, M. & Wink, D. A. (2003) J. Inorg. Biochem. 93, 52–60. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi, A., Suzuki, Y., Kamikawa, T., Hayashi, H. & Yamazakim, N. (1980) Life Sci. 27, 1679–1685. [DOI] [PubMed] [Google Scholar]
- 31.Wink, D. A. & Mitchell, J. B. (1998) Free Radical Biol. Med. 25, 434–456. [DOI] [PubMed] [Google Scholar]
- 32.Bazylinski, D. A. & Hollocher, T. C. (1985) Inorg. Chem. 24, 4285–4288. [Google Scholar]
- 33.Nelli, S., Hillen, M., Buyukafsar, K. & Martin, W. (2000) Br. J. Pharmacol. 131, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wink, D. A., Grisham, M. B., Miles, A. M., Nims, R. W., Krishna, M. C., Pacelli, R., Teague, D., Poore, C. M. B., Cook, J. A. & Ford, P. C. (1996) Methods Enzymol. 268, 120–130. [DOI] [PubMed] [Google Scholar]
- 35.Gbadegesin, M., Vicini, S., Hewett, S. J., Wink, D. A., Espey, M., Pluta, R. M. & Colton, C. A. (1999) Am. J. Physiol. 277, C673–C683. [DOI] [PubMed] [Google Scholar]
- 36.Gratzel, M., Taniguchi, S. & Henglein, A. (1970) Ber. Bunsenges. Phys. 74, 1003–1010. [Google Scholar]
- 37.Shafirovich, V. & Lymar, S. V. (2002) Proc. Natl. Acad. Sci. USA 99, 7340–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paolocci, N., Katori, T., Champion, H. C., St. John, M. E., Miranda, K. M., Fukuto, J. M., Wink, D. A. & Kass, D. A. (2003) Proc. Natl. Acad. Sci. USA 100, 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Espey, M. G., Miranda, K. M., Thomas, D. D. & Wink, D. A. (2002) Free Radical Biol. Med. 33, 827–834. [DOI] [PubMed] [Google Scholar]
- 40.Pittner, R. A., Albrandt, K., Beaumont, K., Gaeta, L. S., Koda, J. E., Moore, C. X., Rittenhouse, J. & Rink, T. J. (1994) J. Cell. Biochem. 55, Suppl. 19–28. [DOI] [PubMed] [Google Scholar]
- 41.Takami, K., Hashimoto, K., Uchida, S., Tohyama, M. & Yoshida, H. (1986) Jpn. J. Pharmacol. 42, 345–350. [DOI] [PubMed] [Google Scholar]
- 42.McEwan, J. R., Ritter, J. M. & MacDermott, J. (1986) Cardiovasc. Res. 23, 921–927. [DOI] [PubMed] [Google Scholar]
- 43.Stanbury, D. M. (1989) Adv. Inorg. Chem. 33, 69–136. [Google Scholar]
- 44.Miranda, K. M., Yamada, K., Espey, M. G., Thomas, D. D., DeGraff, W., Mitchell, J. B., Krishna, M. C., Colton, C. A. & Wink, D. A. (2002) Arch. Biochem. Biophys. 401, 134–144. [DOI] [PubMed] [Google Scholar]
- 45.Bartberger, M. D., Liu, W., Ford, E., Miranda, K. M., Switzer, C., Fukuto, J. M., Farmer, P. J., Wink, D. A. & Houk, K. N. (2002) Proc. Natl. Acad. Sci. USA 99, 10958–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonner, F. T. & Ravid, B. (1975) Inorg. Chem. 14, 558–563. [Google Scholar]
- 47.Zamora, R., Grzesiok, A., Weber, H. & Feelisch, M. (1995) Biochem. J. 312, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janaway, G. A. & Brauman, J. I. (2000) J. Phys. Chem. A 104, 1795–1798. [Google Scholar]
- 49.Bielski, B. H. J., Cabelli, D. E., Arudi, R. L. & Ross, A. B. (1985) J. Phys. Chem. Ref. Data 14, 1071–1072. [Google Scholar]
- 50.Fee, J. A. & DiCorleto, P. E. (1973) Biochemistry 12, 4893–4899. [DOI] [PubMed] [Google Scholar]
- 51.Koppenol, W. H. (1998) Free. Radical Biol. Med. 25, 385–391. [DOI] [PubMed] [Google Scholar]
- 52.Nakano, M., Kimura, H., Hara, M., Kuroiwa, M., Kato, M., Totsune, K. & Yoshikawa, T. (1990) Anal. Biochem. 187, 277–280. [DOI] [PubMed] [Google Scholar]
- 53.Meister, A. & Anderson, M. E. (1982) Annu. Rev. Biochem. 52, 711–760. [DOI] [PubMed] [Google Scholar]
- 54.Liu, X. P., Miller, M. J. S., Joshi, M. S., Thomas, D. D. & Lancaster, J. R. (1998) Proc. Natl. Acad. Sci. USA 95, 2175–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geppetti, P., Tramontana, M., Santicioli, P., Del Bianco, E., Giuliani, S. & Maggi, C. A. (1990) Neuroscience 38, 687–692. [DOI] [PubMed] [Google Scholar]
- 56.Huang, M. H., Knight, P. R., III, & Izzo, J. L., Jr. (1999) Am. J. Physiol. 276, R259–R264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.