

Abstract
Influenza is a paradigm for understanding viral infections. As an opportunistic pathogen exploiting the cellular endocytic machinery for infection, influenza is also a valuable model system for exploring the cell's constitutive endocytic pathway. We have studied the transport, acidification, and fusion of single influenza viruses in living cells by using real-time fluorescence microscopy and have dissected individual stages of the viral entry pathway. The movement of individual viruses revealed a striking three-stage active transport process that preceded viral fusion with endosomes starting with an actin-dependent movement in the cell periphery, followed by a rapid, dynein-directed translocation to the perinuclear region, and finally an intermittent movement involving both plus- and minus-end-directed microtubule-based motilities in the perinuclear region. Surprisingly, the majority of viruses experience their initial acidification in the perinuclear region immediately following the dynein-directed rapid translocation step. This finding suggests a previously undescribed scenario of the endocytic pathway toward late endosomes: endosome maturation, including initial acidification, largely occurs in the perinuclear region.
The infection pathway of influenza is a complex, multistep process: viruses enter cells by receptor-mediated endocytosis, then move from endocytic vesicles to early endosomes and finally to late endosomes where the viruses fuse with the endosomal membrane to release viral genes (1–7), a pathway followed by many other medically important viruses (3, 4). Exploiting the cell's constitutive endocytic machinery for infection, influenza is an ideal probe for exploring the cell's endocytic pathway. However, despite intensive efforts in investigating influenza infection, many critical properties of the endocytosis and endocytic traffic of the virus remain elusive. Among these are important and general questions for cellular endocytosis: what are the transport mechanisms of endocytic compartments at different stages on the endocytic pathway, and how do these compartments and the enclosed endocytic cargo mature (6–8)? The difficulty in real-time imaging of the endocytic pathway presents one of the major hurdles in addressing the above questions. In this work, by tracking the behavior of single influenza viruses in real-time (9–12), we have dissected individual stages of the viral entry process, observed transient, previously unobserved steps in endocytic viral trafficking, and obtained a dynamic picture of the cell's endocytic pathway exploited by influenza. We have discovered that virus-bearing endosomes are transported in cells in three distinct stages, each with a distinct cytoskeleton- and motor-protein-dependent mechanism. We have revealed surprising endocytic acidification dynamics: the initial acidification of endocytic cargo mainly occurs in the perinuclear region, bringing into question the previous picture that early endosomes in the cell periphery are the early acidification sites for endocytic cargo (13, 14).
Materials and Methods
Viruses and Fluorescent Labeling. Influenza virus X-31 was purchased from Charles River Laboratories. To label with lipophilic dyes, the viruses were incubated with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) (Molecular Probes) for 2 h at 22°C. To label with amine-reactive Cy3 and CypHer5 (Amersham Pharmacia), the viruses were incubated with both dyes in a carbonate buffer (pH 9.3) at 22°C for 1 h. In both cases, unbound dye was removed via buffer exchange into 50 mM Hepes buffer (pH 7.4, 145 mM NaCl) using gel filtration columns. Immediately before experiments, viral aggregates were removed with 0.2 μm pore size filters.
Cell Culture and Drug Treatment. Chinese hamster ovary (CHO) cells (American Type Culture Collection) were maintained in a 5% CO2 environment in Dulbecco's modified Eagle medium (DMEM, Invitrogen) with 10% FBS and passaged every 2–3 days. After six to seven passages, the cells were discarded and new aliquots of frozen cells were thawed. For fluorescence imaging, CHO cells were cultured in DMEM with 10% FBS in Petri dishes with poly(l-lysine)-coated glass coverslips on the bottom. Before fluorescence experiments, cells were washed in serum-free, phenol red-free medium fortified with a 100 mM Hepes buffer (pH 8.0). To disrupt microtubules, the cells were incubated with medium containing 60 μM nocodazole for 30 min before experiments. To disrupt actin filaments, the cells were incubated with medium containing 20 μM cytochalasin D (cyto-D) for 30 min before experiments. The drugs were maintained in the cell culture throughout the experiments. To inhibit dynein activity, the cells were microinjected with antidynein intermediate chain mAb (Chemicon) and were allowed to recover in the CO2 incubator for 15 min before experiments. HeLa cells (American Type Culture Collection) were maintained in minimum essential medium (Invitrogen) containing 10% FBS.
Epi-Fluorescence and Differential Interference Contrast (DIC) Imaging. DiD-labeled viruses were excited with a 633-nm helium–neon laser (Melles-Griot), whereas Cy3/Cypher5-labeled viruses were simultaneously excited with a 532-nm diode-pumped Nd:YAG laser (CrystaLaser, Reno, NV) and the helium–neon laser. A custom-designed polychroic beam splitter (Chroma Technology, Brattleboro, VT) that reflects at wavelengths 520–550 nm and 615–655 nm and transmits the rest of the visible spectrum was used to reflect the laser lines onto the sample while transmitting the fluorescent emission. The fluorescent emission was collected by an oil-immersion objective with numerical aperture of 1.45 (Olympus) and imaged onto a charge-coupled device (CCD) camera (Roper Scientific, CoolSnap HQ). The fluorescent emissions from Cy3 and CypHer5 were spectrally separated by 650 nm long-pass dichroic mirrors (Chroma Technologies) and imaged onto two separate areas of the CCD camera. A 585/70-nm band-pass filter was used for Cy3 fluorescent emission, and a 650 long-pass filter was used for CypHer5 emission. Image series were recorded at two frames per second. DIC optics (Olympus) were used to obtain cell images before and after the fluorescence imaging. Experiments were conducted at 37°C.
Image Analysis. Each frame in the movie stack was processed by convolving with a Gaussian spatial filter to remove background and noise (15). Viral peaks were detected by recursively integrating over bright regions connected to each local maximum. The location of each virus was computed as the centroid of the bright region. The peak positions are determined with ≈40 nm accuracy when the viruses are in focus, and the error can be up to 150 nm when the viruses are slightly out of focus. Viral trajectories were reconstructed by pairing peaks in each frame to previously established trajectories according to proximity and similarity in intensity. For quantitative analysis of complete trajectories, only those viruses that start in the peripheral region of cell surfaces and move roughly within the focal plane are analyzed. Viruses binding directly above the perinuclear region tend to move orthogonal to the focal plane. Their transport properties cannot be analyzed quantitatively.
Results and Discussion
Visualizing Infection of Individual Influenza Viruses. First, we investigated the transport and fusion of influenza by tracking single viruses in living cells by using fluorescence microscopy. We used a labeling scheme that allows detection of the membrane fusion of a single virus as well as its physical trajectory. CHO cells and X-31 influenza were used as the model cell and virus systems, respectively. The influenza viruses were labeled with DiD, a fluorescent lipophilic dye that spontaneously partitions into the viral membrane. The dye-labeled viruses are infectious and dye-labeling does not affect the viral infectivity (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org). The surface density of the DiD dye is sufficiently high so that its fluorescence is largely quenched, but still allows single dye-labeled viruses to be clearly detected. A significant increase in fluorescence intensity is expected when viruses fuse with endosomes. This is a commonly used method to signal viral, vesicular, and cellular fusion (16). We measured in vitro the fusion kinetics of labeled viruses with red blood cells at the fusion pH (pH 5) and obtained a fusion rate constant ≈0.1 s–1 (Supporting Text and Fig. 6, which is published as supporting information on the PNAS web site). This result agrees well with previously determined fusion kinetics of influenza viruses or cells expressing influenza fusion proteins both in the presence or absence of the lipophilic dyes (17–19), suggesting that the viral fusion kinetics are not perturbed by the lipophilic dyes.
To illustrate the dramatic difference in the fluorescence signals at different stages of infection, we used a low-temperature preincubation method to allow synchronized infection of many viruses (1). Viruses were incubated with cells at 4°C for 10 min for binding to cells without endocytosis. The temperature was then elevated to 37°C rapidly to initiate infection. Fluorescence images of the viruses at 2 min and 15 min after initiation of infection are shown in Fig. 1a. These images clearly illustrate significant viral transport toward the perinuclear region. Viral fusion with endosomes is evident by fluorescence dequenching. Approximately one-fourth of the viruses had fused after 20 min, consistent with the observation that two-thirds of the viruses were intrinsically fusion-defective (Supporting Text) and, in addition, 15% of the viruses in cells did not reach the perinuclear region within 20 min. In the following real-time virus-tracking experiments, viruses were added to cells either by using the low-temperature preincubation method or in situ at 37°C. The two methods give similar results on the viral transport properties, as well as fusion and acidification kinetics.
Fig. 1.

Fluorescence images and fusion sites of individual influenza viruses in CHO cells. (a) DIC image of a CHO cell (Left) and fluorescence images of DiD-labeled viruses 2 min (Center) and 15 min (Right) after infection. The thin white lines trace the nuclear boundary. (b) The fusion sites of individual viruses are marked by red stars. The boundaries of a cell and its nucleus are highlighted by thick gray and thin white lines, respectively. The boundaries between cells are discernible in the DIC image as distinct cleft-like contours. The nuclei are visible as ovoid regions near the cell centers surrounded by high-contrast vesicular structures. The stars seemingly inside the nuclei most likely indicate fusion events above or below the nuclei.
Three-Stage Active Transport of Viruses in Cells. Tracking individual viruses in real-time allows us to reveal unambiguously the dynamics of viral transport in cells. Fig. 2a shows a typical trajectory of a virus starting in the cell periphery, moving to the perinuclear region and fusing with a late endosome. A movie showing the transport and fusion of this virus is available in Movie 1, which is published as supporting information on the PNAS web site. The time trajectories of the velocity and fluorescence intensity of the virus are shown in Fig. 2b. These results illustrate a previously unknown three-stage transport pattern that is highly reproducible among viruses: the virus first moves slowly in the cell periphery (stage I), then adopts a rapid and unidirectional movement toward the nucleus (stage II), followed by intermittent, often bidirectional movement in the perinuclear region (stage III) before finally fusing with an endosome as indicated by the dramatic fluorescence increase (Fig. 2b). We identify fusion events in single-virus trajectories with step-like, persistent fluorescence increases that are at least a factor of 3, typically more than a factor of 5, above the prefusion intensity. This three-stage transport behavior is critical for influenza infection. Our control experiments with HeLa cells, a cell type known to exhibit inefficient influenza infection (20, 21), show much less viral fusion. Analysis of single viral trajectories in HeLa cells reveals that the majority of viruses are largely limited to the cell periphery without stage II and III transport (Supporting Text and Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 2.

Tracking the transport and fusion of individual influenza viruses. (a) The trajectory of a DiD-labeled virus inside a cell. The color of the trajectory codes time with the colored bar indicating a uniform time axis from 0 s (black) to 500 s (yellow). The red star indicates the fusion site. (Scale bar: 10 μm.) (b) Time trajectories of the velocity (black) and the DiD fluorescence intensity (blue) of the virus. t1, t2, and t3 are the durations of stages I, II, and III, respectively. Stage II movements can be consistently identified for each viral trajectories as the rapid unidirectional translocation from the cell periphery to the perinuclear region. Stage I is then defined as the period before this transient motion, and stage III is defined as the period after stage II but before fusion. (c) Histogram of the viral velocity in stage I. (Inset) Shown is the measured average mean square displacement (〈Δr2〉) vs. time (Δt) for a virus (green symbols). The green line is a fit to 〈Δr2〉 = constant + DΔt + (vΔt)2 with D = 0.001 μm2/s and v = 0.02 μm/s. The small constant term is due to noise. About 60% of the viral trajectories in stage I show such superlinear dependence of 〈Δr2〉 on Δt. Because of the diffusion-like component of the movement (DΔt), the instantaneous speed in the histogram (=Δr/Δt) depends on Δt, which is chosen to be 0.5 s in c–e. (d) Histogram of the viral velocity in stage II. (e) Histogram of the viral velocity in stage III. (Inset) Shown is a typical example of 〈Δr2〉 vs. Δt. To represent the bursts of relatively fast movements in stage III, the 〈Δr2〉 vs. Δt plot was calculated by using only those points where the virus is traveling with speed >0.3 μm/s. The green line is a fit of the first eight data points to 〈Δr2〉 = constant + D′Δt + (v′Δt)2 with D′ = 0.5 μm2/s and v′ = 0.4 μm/s. The small constant term is due to noise.
Actin-Dependent Transport in Stage I. Analysis of single-virus trajectories during these three stages provides further insight into the mechanisms of viral transport. First we characterize stage I, the longest of the three with an average duration of 6 min (Fig. 8a, which is published as supporting information on the PNAS web site). This stage represents movements of viruses occurring in the cell periphery typically within a distance of 2 μm from the site of initial binding. Fig. 2c shows the distribution of instantaneous viral velocities and the dependence of the mean square distance traveled by a virus (〈Δr2〉) on the traveling time (Δt) in this stage. Although free diffusion should lead to a strictly linear dependence of 〈Δr2〉 on Δt, our experimental data clearly show a superlinear dependence with a dominating quadratic term indicating that the viral movement is directed. To test the mechanism associated with this directed movement, we measured the viral trajectories in cells treated with nocodazole, a drug that disrupts microtubules, or with cyto-D, a drug that disrupts actin filaments. The movement observed in nocodazole-treated cells is indistinguishable from the stage I movement in an untreated cell (Fig. 3a). In contrast, the viruses in cyto-D-treated cells show much more limited mobility (Fig. 3a). These results suggest that stage I movement is an actin-dependent active transport. Comparison of the viral trajectories in untreated cells with those in actin-disrupted cells shows that the actin dependent movement starts rapidly, within a few seconds after the virus binds to the cell (Fig. 3b). This movement likely represents the transport of virus-containing endocytic vesicles inside the cell, either directed by myosin motors on actin filaments (7) or propelled by actin filaments in a fashion similar to the actin-based motility of listeria (7, 22, 23). However, we cannot exclude the possibility that stage I involves an actin-dependent motion on the cell surface (24, 25).
Fig. 3.

Effects of drugs and antibodies on viral transport properties. (a) Viral trajectories of the first 100 s of viral movement in untreated, cyto-D-treated, and nocodazole-treated cells. The arrow in Upper Right indicates the minimum radius (rbound) of a circle centered at the origin of the trajectory that can cover the entire trajectory at a given time. (b) The average rbound over many trajectories as a function of time. The rbound in untreated and nocodazole-treated cells quickly exceeds the rbound attained in cyto-D-treated cells. The rapid, initial increase of rbound in the cyto-D-treated curve likely arises from restricted diffusion of the viruses or undulations of the cells, and does not occur for viruses immobolized on glass. (c) Viral trajectories (10–15 min) in untreated cells, nocodazole-treated cells, and cells injected with anti-dynein (1 mg/ml). The time-color scale is similar to that described in Fig. 2. (Scale bar: 10 μm.)
Dynein-Directed Transport on Microtubules in Stage II. Next, we characterize stage II, a transient burst of movement that could only be directly observed with high-speed single-virus imaging. The viral transport in stage II is characterized by a rapid, unidirectional movement from the cell periphery to the perinuclear region with an instantaneous velocity of 1–4 μm/s (Fig. 2 a, b, and d). This value is consistent with the velocity of retrograde dynein (a minus-end-directed motor protein) walking on microtubules (26). This stage II movement typically persists for only a few seconds (Fig. 8b). It is absent from viral trajectories in cells treated with nocodazole (Fig. 3c), indicating that the rapid movement is indeed microtubule-dependent. This stage is also largely inhibited by microinjecting anti-dynein antibodies at 1 mg/ml into the cell (Fig. 3c). The inhibition is less efficient at lower concentrations of anti-dynein (data not shown). These results demonstrate that, in stage II, viruses are transported by dynein motor proteins on microtubules. Because this movement occurs before viral fusion, it represents the movement of the virus-bearing endocytic compartment (endosome or endocytic carrier vesicle; refs. 13 and 27).
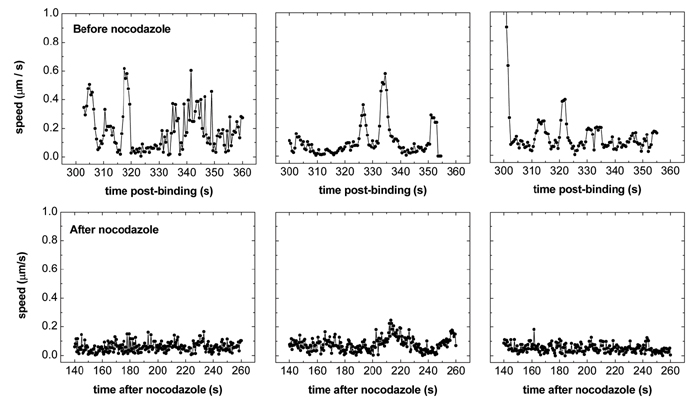
Competing Plus- and Minus-End-Directed Motilities on Microtubules in Stage III. After this rapid stage II movement, the virus experiences a dramatic reduction of velocity and enters stage III in the perinuclear area (Fig. 2 a, b, and e). In this stage, the viruses are observed to move intermittently, often back and forth along the same path, clearly indicating that the movement is directed. The 〈Δr2〉 vs. Δt plot (Fig. 2e) shows a superlinear dependence in the low Δt range consistent with directed movement and an oscillatory behavior in the high Δt range consistent with the back and forth movement. These results strongly suggest that the virus-containing endocytic compartment at this stage is transported by both minus-end-directed motors and plus-end-directed motors on microtubules. The intermittency and relatively low speed of the movement are likely caused by the competition between these motors (11). Indeed, we can selectively block the intermittent stage III movement by adding nocodazole to cells after viruses complete their stage II movement (Supporting Text and Fig. 9, which is published as supporting information on the PNAS web site). The change from unidirectional (stage II) to bidirectional (stage III) movement observed here suggests the presence of a maturation step on the endocytic pathway where the endocytic compartment changes from having predominantly membrane-bound dynein activity to having both minus- and plus-end-directed motor activities, consistent with a previous finding that dynein and kinesin distributions are distinct on different membrane compartments (28).
Viral Fusion. The specific labeling of the virus with membrane dyes allows us to detect not only the physical trajectories but also the fusion events of individual viruses with endosomes, as indicated by fluorescence dequenching (Fig. 2b). The fusion sites are largely in the perinuclear region (Fig. 1b), consistent with viral fusion to late endosomes that are primarily localized near the nucleus (8, 14). The average fusion time obtained from our single-virus trajectories is 8 min after binding, in very good agreement with the previously measured time (8 min) for an ensemble of endocytosed viruses to enter late endosomes in CHO cells (29). This quantitative agreement indicates that dye-labeling does not significantly perturb the viral entry kinetics. The late endosomal pH (pH 5) is critical for influenza fusion (14, 29). Indeed, viral fusion is not observed in the presence of ammonium chloride, a lipophilic weak base known to raise endosomal pH (1) nor is it observed in the presence of nocodazole (data not shown).
It has been suggested that on the endocytic pathway to late endosomes, the endocytosed cargo experiences at least two acidification steps, one from the extracellular pH to the early endosomal pH (pH ≈6) and a later change to the late endosomal pH (pH ≈5) (14, 29–32). Because the late endosomal pH is required for influenza fusion and the fusion kinetics are very rapid at pH 5 (Supporting Text and Fig. 6), viral fusion is an ideal indicator for the second acidification step. Our results show that fusion and thus the second acidification step occurs on average 2 min after the rapid movement in the perinuclear region.
Initial Acidification on the Endocytic Pathway Occurs After Stage II Transport in the Perinuclear Region. We directly probed the early acidification of the endocytic pathway by tracking individual viruses labeled with pH-sensitive dyes. This approach allowed us to simultaneously correlate pH, cellular location and kinematic information of individual viruses. The viruses were co-labeled with pH-sensitive CypHer5 dyes and pH-insensitive Cy3 dyes. This dye-labeling procedure also does not affect the viral infectivity (Supporting Text). The ratio of fluorescent emission from the two dyes is a pH indicator between pH 6 and 8. Fig. 4 a and b show a typical viral trajectory inside the cell and the corresponding time trajectories of viral velocity and pH, respectively. Movie 2, showing the transport and acidification of this virus is published as supporting information on the PNAS web site. Surprisingly, the initial acidification of the virus to pH 6 occurs in the perinuclear region after the rapid stage II movement. Statistics (Fig. 4c) indicate that the majority of viruses experience their initial acidification step after the rapid movement into the perinuclear region. Moreover, the initial acidification time is strongly correlated with the starting time of the rapid movement (Supporting Text and Fig. 10, which is published as supporting information on the PNAS web site). The average time elapsed between rapid movement and the initial acidification is only on average 0.5 min, clearly indicating that this is a distinct step from the acidification that induces fusion (Fig. 4c).
Fig. 4.

Tracking the transport and acidification of individual influenza viruses. (a) The trajectory of a Cy3/CypHer5-labeled virus inside a cell. The colored bar indicates a uniform time axis from 0 to 450 s. The green star indicates the initial acidification site. (Scale bar: 10 μm.) (b) Time trajectories of the velocity (black) and the ratio of CypHer5 and Cy3 fluorescent emission (red) of the virus. t4 defines the time elapsed between the end of stage II and the initial acidification event. The pH values are labeled according to the calibrated pH dependence of the intensity ratio between CypHer 5 and Cy3 (data not shown). (c) Histograms of t3 and t4 as defined in Figs. 2b and 4b, respectively.
The above results paint a striking picture: the majority of endocytic compartments carrying viruses experience their initial acidification in the perinuclear region. This result brings into question a previously suggested picture that early endosomes, which are distributed in the peripheral region of a cell and mature to pH ≈6, are the early acidification site of endocytic cargos (13, 14, 29–32). Instead, our results suggest that the virus-containing endocytic compartments leave early endosomes before the acidification of these endosomes and the acidification of the viral cargo itself occurs largely in the perinuclear region following rapid movement on microtubules. Notably, in cells treated with nocodazole to inhibit the rapid movement, viruses do experience acidification to pH 6 in the cell periphery, but with the acidification slower than in untreated cells (Supporting Text). Thus, the surprisingly late initial acidification step in the perinuclear region indicates a kinetic relationship between the initial acidification and the rapid microtubule-dependent movement in normal cells rather than a requirement of microtubule-dependent movement for the initial acidification.
Conclusion
In summary, we have used real-time fluorescence microscopy to track the infection pathway and dynamics of single influenza viruses in living cells. By using fluorescent indicators to monitor the acidification and fusion of viral particles, we have investigated the relationship between crucial steps on the viral infection pathway and endocytic transport properties at the single-virus level. Our single-virus trajectories clearly dissect the viral endocytic pathway and unambiguously demonstrate that the viral transport is composed of three distinct stages before virus-endosome fusion, with stage I being an actin-dependent active transport in the cell periphery, stage II being a rapid and dynein-directed movement on microtubules toward the perinuclear region, and stage III being an intermittent active transport involving both plus- and minus-end-directed motor proteins on microtubules in the perinulcear region. The single-virus trajectories reveal three maturation steps of the virus-containing endocytic compartments after rapid stage II transport: change of the membrane-bound motor-protein distribution or activity, initial acidification to pH 6, and later acidification to the influenza fusion pH (pH 5). To our surprise, the initial acidification step occurs in the perinuclear region after the rapid movement on microtubules, bringing into question the previous suggestion that early endosomes are the early acidification sites for endocytic cargo targeted to endosomes and lysosomes. Combining previous results (7) and findings from this work, we propose a model of the endocytic pathway toward late endosomes illustrated in Fig. 5. As influenza is an opportunistic pathogen exploiting of the cell's constitutive endocytic pathway for infection, the model derived here (Fig. 5) should have general implications for the cellular endocytic pathway.
Fig. 5.

A model of the endocytic pathway toward late endosomes. (1) The virus is internalized and transported to the early endosome (EE) in an actin (AT)-dependent way (stage I movement). (2) The virus-containing endocytic compartment leaves the EE, still at the extracellular pH. This may occur either through a virus-bearing endocytic carrier vesicle (ECV) budding from the EE (13, 27) or the membrane-rich tubular region of the EE recycling to leave a more vesicular EE that contains the virus (33). (3) The ECV or vesicular EE is transported to the perinuclear region via a dynein-directed movement on a microtubule (MT) (stage II movement). (4) The ECV or vesicular EE matures into a maturing endosome (ME) by changing the membrane-bound motor protein activity (transition from stage II to stage III movement). (5) The endosome further matures by changing its pH from the extracellular value to pH ≈6 (initial acidification as indicated by the change in fluorescence ratio between CypHer 5 and Cy3 conjugated to viruses). (6) Further acidification brings the pH of the endosome to the late endosomal (LE) value, pH ≈5 (second acidification as indicated by viral fusion).
Supplementary Material
Acknowledgments
We thank Judith White, Stephen Harrison, Timothy Mitchison, Kenneth Kosik, and Sunney Xie for helpful comments on the manuscript, and Judith White for suggestions on cell culture. This work was supported in part by the Office of Naval Research (X.Z.). M.J.R. is supported by a predoctoral fellowship from the National Science Foundation.
Abbreviations: CHO, Chinese hamster ovary; DIC, differential interference contrast; cyto-D, cytochalasin D; DiD, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine.
References
- 1.Matlin, K. S., Reggio, H., Helenius, A. & Simons, K. (1981) J. Cell Biol. 91, 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin, K. & Helenius, A. (1990) J. Virol. 65, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helenius, A. & Marsh, M. (1989) Adv. Virus Res. 36, 107–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klasse, P. J., Bron, R. & Marsh, M. (1998) Adv. Drug. Delivery Rev. 34, 65–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein, J. L., Brown, M. S., Anderson, R. G. W., Russell, D. W. & Schneider, W. J. (1985) Annu. Rev. Cell Biol. 1, 1–39. [DOI] [PubMed] [Google Scholar]
- 6.Gruenberg, J. (2001) Nat. Rev. Mol. Cell Biol. 2, 721–730. [DOI] [PubMed] [Google Scholar]
- 7.Apodaca, G. (2001) Traffic 2, 149–159. [DOI] [PubMed] [Google Scholar]
- 8.Helenius, A., Mellman, I., Wall, D. & Hubbard, A. (1983) Trends Biochem. Sci. 8, 245–250. [Google Scholar]
- 9.Georgi, A., Mottola-Hartshorn, C., Warner, W., Fields, B. & Chen, L. B. (1990) Proc. Natl. Acad. Sci. USA 87, 6579–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seisenberger, G., Ried, M. U., Endreb, T., Buning, H., Hallek, M. & Brauchle, C. (2001) Science 294, 1929–1932. [DOI] [PubMed] [Google Scholar]
- 11.Suomalainen, M., Nakano, M. Y., Keller, S., Boucke, K., Stidwill, R. P. & Greber, U. F. (1999) J. Cell Biol. 144, 657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald, D., Vodicka, M. A., Lucero, G., Svitkina, T. M., Borisy, G. G., Emerman, M. & Hope, T. J. (2002) J. Cell Biol. 159, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bayer, N., Schober, D., Prchla, E., Murphy, R. F., Blaas, D. & Fuchs, E. (1998) J. Virol. 72, 9645–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellman, I., Fuchs, R. & Helenius, A. (1986) Annu. Rev. Biochem. 55, 663–700. [DOI] [PubMed] [Google Scholar]
- 15.Russ, J. C. (1999) in The Image Processing Handbook, ed. Whitehead, C. (CRC Press, Boca Raton, FL), pp. 227–303.
- 16.Loyter, A., Citovsky, V. & Blumenthal, R. (1988) Methods Biochem. Anal. 33, 129–164. [DOI] [PubMed] [Google Scholar]
- 17.Stegmann, T., Morselt, H. W. M., Scholma, J. & Wilschut, J. (1987) Biochim. Biophys. Acta 904, 165–170. [DOI] [PubMed] [Google Scholar]
- 18.Clague, M. J., Schoch, C. & Bluementhal, R. (1991) J. Virol. 65, 2402–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spruce, A. E., Iwata, A., White, J. M. & Almer, W. (1989) Nature 342, 555–558. [DOI] [PubMed] [Google Scholar]
- 20.Gujuluva, C. N., Kundu, A., Murti, K. G. & Nayak, D. P. (1994) Virology 204, 491–505. [DOI] [PubMed] [Google Scholar]
- 21.Lohmeyer, J., Talens, L. T. & Klenk, H. D. (1979) J. Gen. Virol. 42, 73–88. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg, M. B. (2001) Microbiol. Mol. Biol. Rev. 65, 595–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merrifield, C. J., Moss, S. E., Ballestrem, C., Imhof, B. A., Giese, G., Wunderlich, I. & Almers, W. (1999) Nat. Cell Biol. 1, 72–74. [DOI] [PubMed] [Google Scholar]
- 24.Basciano, P. A. & King-Smith, C. (2002) Pigment Cell Res. 15, 184–191. [DOI] [PubMed] [Google Scholar]
- 25.Theriot, J. A. & Mitchison, T. J. (1992) J. Cell Biol. 118, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma, S. & Chrisholm, R. L. (2002) J. Cell Sci. 115, 1453–1460. [DOI] [PubMed] [Google Scholar]
- 27.Gruenberg, J. & Howell, K. E. (1989) Annu. Rev. Cell Biol. 5, 453–481. [DOI] [PubMed] [Google Scholar]
- 28.Lin, S. X. H., Pfister, K. K. & Collins, C. A. (1996) Cell Motil. Cytoskel. 34, 299–312. [DOI] [PubMed] [Google Scholar]
- 29.Schmid, S., Fuchs, R., Keilian, M., Helenius, A. & Mellman, I. (1989) J. Cell Biol. 108, 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ichimura, T., Hatae, T. & Ishida, T. (1997) Eur. J. Cell Biol. 74, 41–48. [PubMed] [Google Scholar]
- 31.Kielian, M. C., Marsh, M. & Helenius, A. (1986) EMBO J. 5, 3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmid, S., Fuchs, R., Male, P. & Mellman, I. (1988) Cell 52, 73–83. [DOI] [PubMed] [Google Scholar]
- 33.Mayor, S., Presley, J. F. & Maxfield, F. R. (1993) J. Cell Biol. 121, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}