

Abstract
This paper critically reviews examples in the literature of photochemical cis-trans isomerization paying particular attention to the medium effect and accompanied conformational changes. A case is made that the Hula-Twist mechanism, postulated in 1985 as a photochemical reaction pathway for a polyene chromophore imbedded in a protein binding cavity such as those of rhodopsin and bacteriorhodopsin, is also a dominant reaction pathway for a diene, or a longer polyene confined in a rigid (relative to isomerization rate) medium. The conventional one-bond-flip process is the preferred reaction pathway in a fluid medium. While defining experiments are proposed, this dual mechanistic approach successfully accounts for all examples in the literature on photoisomerization reactions whether involving conformational changes or not.
Photochemistry of conjugated
polyenes has been the subject of considerable study (1–3). Despite the
relative simplicity of the molecules, the results have been quite
instructive. One important lesson, pointed out by Havinga and
Schlatmann (4) and subsequently elaborated by other researchers (3, 5)
through their detailed study of the photochemistry of polyenes in the
vitamin D series (electrocyclizations, sigmatropic rearrangements, and
geometric isomerizations) was the concept
nonequilibration of excited
rotamers, later popularly known as the NEER Principle (3,
5).§ In an incisive discussion, they pointed out that the
results indicated that the excited molecules apparently do not undergo
rotation about single bonds that connect unsaturated centers in the
ground states, a reasonable hypothesis because theory indicated that
these linkages greatly have increased double bond character in π,π*
excited states. The NEER effect is illustrated with reactions of
cis-1,3,5-hexatriene (1) (3, 5).
![]()
We showed that the excited triplet states of the simple dienes must
retain the ground state orientation about the central single bonds (6,
7). This was an especially telling case because the excited species
even lived long enough to undergo bimolecular reactions in solution.
The case of the parent 1,3-butadiene (2)
is shown.¶
![]()
Subsequently, there have been a large number of confirmatory reports of the same effect both from Leiden and from many other laboratories. Space limitation does not allow us to review a majority of these examples. Instead, we shall mention a few major developments germane to the current discussion. Isomerization reaction of substituted dienes (whether excited singlets or triplets) exhibits reactivities specific to different excited conformers (8). Different spectra for isomeric triplets have been reported (9). Substituent- and wavelength-dependent photochemistry of dienes and trienes has been shown to be consistent with selective excitation of different conformers (5, 10). And, from a variety of arylalkenes, where fluorescence is more common or efficient than in the simple dienes and trienes, different excited-state properties of the isomeric excited singlets have been demonstrated (11–13). For longer polyenes, the effort has been largely devoted to unravelling the complex chemistry associated with the large number of geometric isomers (14, 15). However, selective excitations of conformers were implicated (16). In short, there have been an overwhelmingly large number of observations in agreement with NEER.
Geometric isomerization is an important mode of reaction of polyenes in their excited singlet states. The prevailing view (1, 8) for this simple reaction is torsional relaxation of the excited double bond of reduced bond order. Crossing to the ground state occurs near the perpendicular geometry. From there on, continuation of the torsional relaxation completes the diabatic reaction. For designating this general mechanistic process, we shall use the term one-bond-flip (OBF) to emphasize the turning over of one half of the molecule during the isomerization process. In 1985, R.S.H.L. proposed a volume-conserving cis-trans isomerization mechanism (17) that was suggested as the primary photochemical process for a confined polyene such as the retinyl chromophore in the visual pigment rhodopsin. This process does not involve turning over either half of the polyene, but instead translocates a single C-H unit by simultaneously rotating two connected single and double bonds. Hence, the name concerted-twist at center n (CT-n) first was introduced. But subsequently, a locally fetching term, Hula-Twist at center n (HT-n), has taken over in popularity (18). In Fig. 1, we show the HT-n and the OBF mechanisms in photoisomerization of a segment of a polyene unit.
Figure 1.

Cis-trans isomerization of a segment of a polyene. (Upper) The Hula-Twist (HT) process showing the translocation of one C-H unit and the sliding sideways movement of the remaining polyene chain. (Lower) The conventional one-bond-flip (OBF) process showing the turning over of one-half of the molecule.
In addition to the difference in volume demand for the two processes (vide infra), the stereochemical consequences also are different. The OBF process leads to isomerization at the reacting double bond only, whereas the HT-n process leads additionally to conformational isomerization at the adjacent single bond. The latter change is seemingly a non-NEER behavior. In an effort to seek a definitive explanation for all documented examples of such apparent non-NEER behavior, we arrived at a unifying view for photoisomerization. We believe there is sufficient literature indicating the validity of this idea.
Examples in Apparent Violation of NEER.
There are several examples in the literature where UV irradiation led
to conformational changes of a diene or polyene unit, in apparent
“violation” of NEER although none ever were discussed in that
light. Thus, Squillacote et al. (19) showed that
S-cis and S-trans
1,3-butadiene (2) trapped in argon matrix at 15 K
interconverted readily on UV irradiation. Subsequently, Squillacote
et al. (20, 21) extended similar studies to isoprene
(2-methylbutadiene), 2,3-dimethylbutadiene, and other alkylated dienes.
Independently, Kohler and coworkers (22, 23) reported that irradiation
of all-trans-1,3,5,7-octatetraene
(3) in an n-octane matrix at
4.2 K led to reversible formation of its 2-S-cis
conformer.
![]() These works stand out in disharmony with the reported unique
chemistry of different excited rotamers (8). More recently, Fuss and
coworkers (24) conducted a low temperature (liquid nitrogen)
photochemical study of two conformers of previtamin D (4 a
and b), and reported the exclusive formation of
photoproducts (5 a and b) derived from
simultaneous twisting of a double bond and an adjacent single bond.
These works stand out in disharmony with the reported unique
chemistry of different excited rotamers (8). More recently, Fuss and
coworkers (24) conducted a low temperature (liquid nitrogen)
photochemical study of two conformers of previtamin D (4 a
and b), and reported the exclusive formation of
photoproducts (5 a and b) derived from
simultaneous twisting of a double bond and an adjacent single bond.
These are unambiguous examples of photochemical
reactions involving conformational changes, in apparent violation of
NEER.
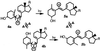 ‖ The modes
of transformation, however, are consistent with the volume-conserving
HT-n mechanism for cis-trans isomerization. In fact, the butadiene and
octatetraene examples were cited as supporting evidence for the HT-n
mechanism (17) and the photochemical results of previtamin D were
labeled as the first definitive example of HT-n (24).
‖ The modes
of transformation, however, are consistent with the volume-conserving
HT-n mechanism for cis-trans isomerization. In fact, the butadiene and
octatetraene examples were cited as supporting evidence for the HT-n
mechanism (17) and the photochemical results of previtamin D were
labeled as the first definitive example of HT-n (24).
Other recent indirect examples in support of HT-n are the following. In
an elegant study with 1,4-dideutero-2,3-dimethylbutadiene
(6) (16), Squillacote and Semple (20) reported the low
efficiency of 2,3-dimethylbutadiene in undergoing S-cis/S-trans
interconversion.
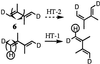
They concluded that for this compound, the preferred chemical
channel of deactivation is OBF at the 1,2-center. However, it occurred
to us that the combined observations on several dienes studied by
Squillacote et al. (19–21) can be consistently accounted
for by the HT-n mechanism. With the presence of a smaller C-H unit at
C-2 or C-3 of a 1,3-diene unit [C-3 for isoprene, 2-isopropylbutadiene
(7), and 2,4-hexadiene
(8)], HT-n should proceed with equal
ease, resulting in the observed interconversion of conformers.
![]() When a
larger substituent is present (such as a
methyl),** steric inhibition makes HT-n
at such centers competitively unfavorable as compared with HT-n at the
unsubstituted terminal centers.
When a
larger substituent is present (such as a
methyl),** steric inhibition makes HT-n
at such centers competitively unfavorable as compared with HT-n at the
unsubstituted terminal centers.
It should be noted that the latter HT-n processes (the translocation of either the C-H or the C-D unit) will achieve the same stereochemical consequence as the proposed (21) OBF mechanism for 6 (the translocation of both the C-H and the C-D unit). In consideration of photoisomerization efficiencies of a series of exocyclic dienes, Leigh and Postigo (28) noted a preference for cases where an adjacent single bond is twisted. Along with the HT-n mechanism, they cited the theoretical studies that traced the preferred relaxation pathways of an excited conjugated hydrocarbon (such as octatetraene; refs. 29 and 30) through several conical intersections which assume geometries corresponding to simultaneous twisting of an adjacent pair of double and single bonds. And a recent molecular modeling study describing the deactivation pathway of the excited rhodopsin (31) showcased HT-12 (for more details, see Protein-Bound Polyene Chromophores) as the primary photochemical reaction.
A Unifying Explanation.
We would like to offer a unifying explanation for the seemingly contradictory photoisomerization results. We first shall concentrate our discussion on two systems where there are documented information for both retention and loss of conformational integrity. Specifically, we wish to address the following two questions. (i) Why did Fuss and coworkers (24) observe the two-bond twist result with two trienes in the vitamin D series (24), whereas nearly all others (3, 5) reported retention of conformational integrity of trienes? (ii) Why were butadiene (19) and octatetraene (22) shown spectroscopically to undergo conformational equilibration although such conformational scrambling was not detected in many others' careful chemical work of substituted dienes (8)? The answer becomes evident if one focuses on the fact that all examples involving conformational changes were carried out in frozen solid media, whereas nearly all photochemial studies were carried out in fluid solutions. The very different reaction conditions present the very likely possibility of the involvement of two different reaction mechanisms.
For molecules in fluid solutions, the conventional OBF mechanism should apply, i.e., the reduced excited-state bond order of the formal double bond permits rapid torsional relaxation to the perpendicular structure. Crossing to the ground state potential surface followed by torsional relaxation completes the diabatic process. The fact that the NEER behavior has operated to near perfection in literature examples is a reflection of the importance of relative excited-state bond orders in dictating the position of the initial twisting process. However, it also is known that this facile torsional motion is highly sensitive to medium viscosity effects. Thus, the efficient isomerization reaction and the weak fluorescence of trans-stilbene observed in fluid solutions can be reversed in rigid media and/or at low temperatures (32–34). Similarly, fluorescence yields and excited-state lifetimes of trienes are enhanced greatly in solid solutions at low temperatures (35). Closing the chemical channel of torsional relaxation should give the molecule time to explore other vibrational modes including more complex motions such as that required to do the Hula-Twist.
Thus, we wish to postulate that HT-n is an improbable high-energy process, not favorable in fluid solutions. However, it might become the dominant process in circumstances (solid medium) where the more probable OBF process is prohibited. The structures in Fig. 1 show the different spatial requirements between HT-n and the conventional OBF process. HT-n involves the translocation of a single H-atom (less volume demanding), whereas the conventional OBF process requires the turning over of one-half of the molecule (more volume demanding). It is possible that HT-n cannot be spared from medium viscosity effects, but certainly much less importantly than the OBF process. Thus, not surprisingly Squillacote and Semple (20) observed steric inhibition when the two key vinyl H's were replaced by methyl groups. Secondarily, we note that the motion of the Cn-H unit is accompanied by a sliding (nearly in the plane of the original π-system) motion of the two remaining halves of the molecule. This sliding motion could also be sensitive to medium viscosity effects to the extent that a large interference could render HT-n impossible. Hence, in our mind, the observations by Squillacote et al. (19–21), Kohler et al. (22, 23), and Fuss and coworkers (24) were the consequence of clever usage of the medium allowing revelation of an improbable process.
An alternative way of viewing the HT-n process was suggested to us,‡‡ i.e., rotation of the formal double bond in the excited state to the perpendicular structure, then twisting an adjacent formal single bond in the ground state followed by torsional relaxation of the doubly twisted species. We do not believe that this stepwise mechanism is in agreement with known facts. Medium viscosity effects are expected to have a profound influence only at the early part of the rotational motion. This portion of the twisting motion is identical in the stepwise mechanism and in OBF—i.e., equally inhibited in viscous media. Therefore, there is no reason to suspect that the stepwise mechanism would become more favored than OBF in frozen media. Also, the results of Fuss and coworkers (24) showed that the two-bond-flip product was formed in the absence of any one-bond-twist product, a feature more consistent with the concerted HT-n mechanism than the stepwise mechanism. And the recent calculations on minimum energy pathways for deactivation of excited dienes and polyenes (29, 30) showed that, even in isolation, two-bond twist is not a prohibitively high-energy pathway. In short, we believe HT-n is a concerted diabatic process, higher in energy than OBF, yet becoming observable when the latter is suppressed in confined media. In fact, media constriction might lead to development of new vibrational modes especially appropriate for involvement in the conversion of electronic energy to vibrational energy in the internal conversion process (36).
The proposed explanation is closely analogous to known examples in spectroscopy and even in photochemistry. That a nonfluorescent compound becomes fluorescent in a frozen medium is clearly the result of shutting off of a rapid radiationless deactivation process or processes. And new modes of chemical reactions (sometimes exclusively) detectable only in a frozen medium again require closing of the normal chemical channel(s) in solution. Particularly relevant are the many examples of medium directed regiospecific or stereospecific photochemical reactions in the solid state (37, 38). In this regard, one might expect HT to be of slow rate but not necessarily a low quantum yield process. The latter is because of the likely absence of other efficient competing processes in frozen confined media. Thus, we believe the high quantum yield of isomerization of rhodopsin (0.67; ref. 39) is likely a unique two-bond-twist pathway guided by the protein residues that constitute the highly specific (40) chiral-binding cavity of the visual protein (41).
It is interesting to note that even in solution, solvent viscosity has a definite influence on configurational isomerization reactions. A relevant example is medium-directed regioselective isomerization of compounds in the vitamin A series (42). Conformational isomerization is also highly sensitive to medium/temperature effects. For example, Kohler et al. (22, 23) reported that 2-S-cis-octatetraene reverts to the all-trans-S-trans conformer at temperatures above 50 K, a remarkably fast process. On the other hand, the primary, bulkier S-cis-photoproducts in the vitamin D series were reported to be stable at liquid nitrogen temperatures (24). Clearly, the exact conditions for detecting the primary photoproduct are important.
Returning to the issue of whether HT violates NEER, it should be noted that the observation of new conformation in the product structures is in no way a guarantee that the molecule has undergone “equilibration of excited rotamers.” HT-n, as we discussed above, is a diabatic process, whereas the NEER behavior is strictly an adiabatic phenomenon (43). Any association of HT-n with violation of NEER is likely the result of misconception derived from the method of analysis—ground state product structure analyses for the understanding of an excited phenomenon, a method used almost exclusively by Havinga and coworkers (3, 5) in formulating the NEER concept.
Proposed Experiments.
After the explanation offered above, we are now in a position to define new experiments for the purpose of distinguishing between the HT-n and the OBF mechanisms. And information for finer details about HT-n also can be obtained with carefully designed systems. These are discussed separately below.
The HT-n and the OBF Mechanisms.
The desirable compounds for distinguishing between these two
mechanistic pathways are the following. For 1,4-dideutero-1,3-butadiene
(9), HT-n is expected
to yield the 1-cis-2-S-cis isomer (by
way of HT-2) or the 2-cis isomer (HT-1) as the primary
photoproducts, whereas OBF will give the latter only. For
1,4-dideuteroisoprene (10),
HT-n will give primarily the
2-S-cis-3-cis isomer (HT-3).
![]()
![]() The
1-cis-2-S-cis isomer (HT-2) is not
expected to form easily because of inhibition by the methyl group.
Additionally, the 1-cis and 3-cis isomers (from
HT-1 and HT-4) also will be formed. On the other hand, OBF will give
only the 1-cis and the 3-cis isomers. Probably
the former is slightly favored because of the documented preference of
isomerization at the trisubstituted center over the disubstituted (44).
For the ring-appended diene
11, HT-n will give the
S-cis conformer, whereas OBF will not lead to any detectable
reactions. On the other hand, compounds 12 a and
b cannot undergo HT-n, whereas OBF is
unaffected.
The
1-cis-2-S-cis isomer (HT-2) is not
expected to form easily because of inhibition by the methyl group.
Additionally, the 1-cis and 3-cis isomers (from
HT-1 and HT-4) also will be formed. On the other hand, OBF will give
only the 1-cis and the 3-cis isomers. Probably
the former is slightly favored because of the documented preference of
isomerization at the trisubstituted center over the disubstituted (44).
For the ring-appended diene
11, HT-n will give the
S-cis conformer, whereas OBF will not lead to any detectable
reactions. On the other hand, compounds 12 a and
b cannot undergo HT-n, whereas OBF is
unaffected.
![]()
For compound 13 in the triene series [and
the analogous compound 14 in the stilbene series or many
others where solution studies are known (11)], HT will give the less
stable cis-S-cis isomer, whereas OBF
will give the more stable
all-S-trans-cis isomer.
![]() Parallel
studies with deuterated 1,3,5,7-octatetraene, or other labeled isomers,
could yield equally interesting information on the longer polyene.
Parallel
studies with deuterated 1,3,5,7-octatetraene, or other labeled isomers,
could yield equally interesting information on the longer polyene.
It is clear that for detection of HT-n products, restricted medium must be used and for detection of OBF products fluid solutions should be used. Whether the two mechanisms could be competing against each other in solvents of intermediate viscosity (or other restraining forces) is an interesting situation to be explored. Steady-state irradiation of samples in solids at a low temperature not only has the advantage of possibly eliminating any of the OBF products but also allows accumulation of the unstable S-cis products for structural analyses by vibrational or other less sensitive but informative spectroscopic techniques. Fast time-resolved spectroscopic methods when applied to fluid samples are not suitable substitution for detection of HT-n processes.
Finer Details of HT-n.
The following compounds are likely to yield new information concerning
finer details of HT-n. Compound 15 will
not provide distinguishable chemistry between HT-n and OBF.
![]() However,
when one of the vinylic positions is deuterated (15a),
relative ease of HT-n at the two vinylic centers will be of great
interest. A favorable deuterium isotope effect
(kD/kH > 1) reflecting
enhanced HT-n by deuterium substitution, is in agreement with the
notion that the C-D vibrational amplitude is smaller than that of C-H.
An unfavorable deuterium isotope effect
(kD/kH < 1) could
suggest instead the importance of vibrational (vinyl CH or CD)
electronic coupling in determining rates of radiationless deactivation
(36). If there is a worry of how to distinguish between a possible
competing secondary deuterium isotope effect for the OBF process, one
could use 3deutero-2,4-hexadiene instead
(16).
However,
when one of the vinylic positions is deuterated (15a),
relative ease of HT-n at the two vinylic centers will be of great
interest. A favorable deuterium isotope effect
(kD/kH > 1) reflecting
enhanced HT-n by deuterium substitution, is in agreement with the
notion that the C-D vibrational amplitude is smaller than that of C-H.
An unfavorable deuterium isotope effect
(kD/kH < 1) could
suggest instead the importance of vibrational (vinyl CH or CD)
electronic coupling in determining rates of radiationless deactivation
(36). If there is a worry of how to distinguish between a possible
competing secondary deuterium isotope effect for the OBF process, one
could use 3deutero-2,4-hexadiene instead
(16).
![]() For this compound, a deuterium
isotope effect on HT-3 and HT-4 would show unequal amounts of the
2-cis-3-S-cis and
4-cis-3-S-cis isomers, whereas OBF
will have minimal isotope effect to give the 2-cis or
4-cis products (also obtainable by HT-2).
For this compound, a deuterium
isotope effect on HT-3 and HT-4 would show unequal amounts of the
2-cis-3-S-cis and
4-cis-3-S-cis isomers, whereas OBF
will have minimal isotope effect to give the 2-cis or
4-cis products (also obtainable by HT-2).
If HT-n is inhibited (not eliminated) with the replacement of the vinyl
H's by methyl groups (21), it will be of interest to examine the
effect of other groups such as F and Cl (17,
18) replacing vinyl H's.
![]() One could then ask
questions such as those regarding medium polarity effect on the HT-n of
compounds containing such electronegative substituents.
One could then ask
questions such as those regarding medium polarity effect on the HT-n of
compounds containing such electronegative substituents.
With dienes where the terminal carbons are substituted with alkyl groups larger than the methyl (19), one should be able to determine whether in HT steric inhibition could play a role in determining relative ease in the sideways sliding motion of the alkyl side chains (Fig. 1). This study should be carried out in conjunction with a thorough study of medium effects on HT-n. The work by Fuss and coworkers (24) should be extended from liquid nitrogen temperature to 4 K for detection of medium-imposed barriers to HT-n. Also, even though Squillacote et al. (21) reported no significant difference in the photochemistry of 1,4-dideutero-2,3-dimethylbutadiene in argon and in isopentane at 15 K, a more comprehensive study of medium effect should be conducted.
The above discussion has been limited largely to conjugated dienes, trienes, and tetraenes. We would like to point out that among the voluminous reports on the photochemistry and photophysics of stilbenes and larger arylalkenes (11), selected systems had been examined in solid solution at low temperature. Fischer and coworker (45, 46) and Alfimov et al. (47) independently found an elegant method to freeze-in nonequilibrium trans rotamer mixtures by low-temperature irradiation of the corresponding cis isomers, by using the information that φ(c→t) is less sensitive to solvent viscosity effect than is φ(t→c). The product rotamer composition was believed to be governed by its equilibrium value in the cis form (11, 45, 46). To us, these results are accountable by the involvement of the HT-n mechanism for the nonplanar cis isomers, which are loosely enclosed by solvent molecules, possibly a requirement for reaction for these large molecules. The relative amounts of the conformers of the trans product will depend not only on the equilibrium concentration and the relative absorbance of various conformers of the cis isomer, but also on the relative quantum yields of HT-n for these conformers. That the trans isomers fail to undergo isomerization to the cis isomer is likely a reflection of steric inhibition of a tightly fit solvent cage around the planar trans isomer. In this regard, the recent HT-n work by Fuss and coworkers (24) was carried out, quite interestingly, with the nonplanar cis isomer. Future studies with carefully designed systems (including the use of cis isomers with fused rings) will allow differentiation of the proposed HT-n mechanism against what is currently poorly defined in the literature.
Many of the proposed experiments are beyond the capability of the facilities available in Hawaii. It is our hope that this paper will provide sufficient stimulation to many experts around the world who have the necessary tools to search for more examples associated with the phenomenon described here—especially because conditions for HT-n now are well defined.
Protein-Bound Polyene Chromophores.
The HT-n concept was introduced to account for the rapid isomerization
reaction of the anchored, excited retinyl chromophore confined in the
visual protein opsin (17). When taken into consideration of possible
isomerization of rhodopsin (11-cis) (20) and
isorhodopsin (9-cis) (21) to apparently a common
primary photoproduct (48), R.S.H.L. postulated HT-10
and HT-11 as the respective primary photochemical processes for these
pigments (17).
![]() However, because of subsequent observation of
photoreactivity of the rhodopsin analog 22
(49–51), the new mode of HT-12 was invoked (49).
However, because of subsequent observation of
photoreactivity of the rhodopsin analog 22
(49–51), the new mode of HT-12 was invoked (49).
![]() Ishiguro (31)
recently demonstrated by molecular modeling that HT-12 is the preferred
photoisomerization process of rhodopsin giving
12-S-cis intermediate 23. However, we
are no longer convinced that for a tethered and anchored chromophore
such as that in rhodopsin, the media and molecular constraints will
necessarily allow the HT-n process to reach an energy well
corresponding to stable S-cis conformation
sufficiently deep for its detection. Recognizing the possibility
of secondary reactions is a departure from our original conjecture
(17).
Ishiguro (31)
recently demonstrated by molecular modeling that HT-12 is the preferred
photoisomerization process of rhodopsin giving
12-S-cis intermediate 23. However, we
are no longer convinced that for a tethered and anchored chromophore
such as that in rhodopsin, the media and molecular constraints will
necessarily allow the HT-n process to reach an energy well
corresponding to stable S-cis conformation
sufficiently deep for its detection. Recognizing the possibility
of secondary reactions is a departure from our original conjecture
(17).
For bacteriorhodopsin, a HT-n process is possibly taking place as well for the conversion of its all-trans retinyl chromophore to the 13-cis isomer. Schulten (52) was the first one who suggested the 13,14-dicis structure for K intermediate, a feature consistent with the HT-14 process (53). Schulten subsequently reported additional evidence in support of the dicis structure (54). This conclusion is different from the 14-trans structure reached after vibrational analyses (55).
For the more recently discovered photoactive yellow pigment (PYP), high-resolution protein structure (0.85 Å) is now available (56). It is interesting to note that a recently proposed structure of the primary product, the red-shifted intermediate (57), is indeed one closely resembling that predicted from a HT process.
In fact, for any other photosensitive polyene pigments (such as bilirubin and phytochrome; ref. 58) one should ask the same question, how isomerization of the imbedded chromophore can take place at an extremely fast rate.
In summary, that HT-n is a high-energy process observable only in restricted medium was the theme expressed in the original HT-n paper (17). Literature evidence now suggests that the same situation applies to small molecules. This theme is different from that of Fuss and coworkers (24) who, in reporting the first example of HT-n, suggested that HT-n could be a common mechanism for all isomerization reactions. We believe that photoisomerization data currently available in the literature can be successfully accounted for by the dual mechanistic approach described in this paper.
Acknowledgments
We thank Professor Jack Saltiel for his critiques to the first paper on HT-n and a first version of this paper. The unique environment in Hawaii helped R.S.H.L. to crystallize the simple ideas reported here. We also wish to express our fond Aloha to all those who nurtured our photochemical thought processes in the past decades. Corey Liu contributed considerable time in reformatting the manuscript into the final form. The current Grants that partially supported this paper are National Institutes of Health DK-17806 and Army Research Office DAAD 19-99-0295.
Abbreviations
- HT-n
Hula-Twist at center n
- NEER
nonequilibrating excited rotamers
- OBF
one-bond-flip
Footnotes
It occurred to us that the NEER phenomenon, although general in scope, is not of the nature of a fundamental law or doctrine proven beyond doubt. It seems that the use of the NEER effect is more appropriate for the phenomenon. In fact, NEER assumption and NEER hypothesis have appeared in the literature (11).
The diradical structure is a simplified representation for equilibrating methyleneallyl radicals shown in sensitized isomerization studies (8).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.210323197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.210323197
Hence, a large okole, the Hawaiian word for “hips.”
A few other exceptions to NEER can be attributed to the long triplet lifetimes that allowed equilibration of triplet conformers. But the fact that, in these cases, the excitations are localized in a large aryl portion of the chromophore makes them incongruent with the original spirit of NEER (25–27).
Professor J. Saltiel expressed this view to us soon after publication of our 1985 HT-n paper (17). The issue was revisited recently along with new examples of NEER behavior.
References
- 1.Saltiel J, Sears D F, Jr, Ko D-H, Pake K-M. In: Handbook of Photochemistry and Photobiology. Horspool W M, Song P-S, editors. Boca Raton, FL: CRC; 1995. pp. 3–15. [Google Scholar]
- 2.Leigh W. In: Handbook of Photochemistry and Photobiology. Horspool W M, Song P-S, editors. Boca Raton, FL: CRC; 1995. pp. 123–142. [Google Scholar]
- 3.Laarhoven W H, Jacobs H J C. In: Handbook of Photochemistry and Photobiology. Horspool W M, Song P-S, editors. Boca Raton, FL: CRC; 1995. pp. 143–154. [Google Scholar]
- 4.Havinga E, Schlatmann J L M A. Tetrahedron. 1961;16:146–152. [Google Scholar]
- 5.Jacobs H J C, Havinga E. Adv Photochem. 1979;11:305–373. [Google Scholar]
- 6.Hammond G S, Liu R S H. J Am Chem Soc. 1963;85:477–478. [Google Scholar]
- 7.Liu R S H, Turro N J, Hammond G S. J Am Chem Soc. 1965;87:3406–3412. [Google Scholar]
- 8.Saltiel J, D'Agostino J, Megarity E D, Metts L, Neuberger K R, Wrighton M, Zafiriu O C. Org Photochem. 1973;3:1–113. [Google Scholar]
- 9.Langkilde F W, Jensen N-H, Wilbrandt R W, Brouwer A M, Jacobs H J C. J Phys Chem. 1987;91:1029–1041. [Google Scholar]
- 10.Dauben W G, Rabinowitz J, Vietmeyer N D, Wendschuh P H. J Am Chem Soc. 1972;94:4285–4292. [Google Scholar]
- 11.Mazzucato U, Momicchioli F. Chem Rev (Washington, DC) 1991;91:1679–1719. [Google Scholar]
- 12.Saltiel J, Tarkalanov N, Sears D F. J Am Chem Soc. 1995;117:5586–5587. [Google Scholar]
- 13.Saltiel J, Zhang Y, Sears D F. J Am Chem Soc. 1997;119:11202–11210. [Google Scholar]
- 14.Ganapathy S, Liu R S H. Photochem Photobiol. 1992;56:959–964. [Google Scholar]
- 15.Saltiel J, Crowder J M, Wang S. J Am Chem Soc. 1999;121:895–902. [Google Scholar]
- 16.Ganapathy S, Liu R S H. J Am Chem Soc. 1992;114:3459–3464. [Google Scholar]
- 17.Liu R S H, Asato A E. Proc Natl Acad Sci USA. 1985;82:259–263. doi: 10.1073/pnas.82.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nickon A, Silversmith E F. Organic Chemistry: The Name Game. New York: Pergamon; 1987. pp. 292–294. [Google Scholar]
- 19.Squillacote M E, Sheridan R S, Chapman O L, Anet F A L. J Am Chem Soc. 1979;101:3657–3659. [Google Scholar]
- 20.Squillacote M E, Semple T C. J Am Chem Soc. 1990;112:5546–5551. [Google Scholar]
- 21.Squillacote M E, Semple T C, Mui P W. J Am Chem Soc. 1985;107:6842–6846. [Google Scholar]
- 22.Akerman J R, Forman S A, Hossain M, Kohler B. J Chem Phys. 1984;80:39–44. [Google Scholar]
- 23.Kohler B. Chem Rev. 1993;93:41–54. [Google Scholar]
- 24.Muller A M, Lochbrunner S, Schmid W E, Fuss W. Angew Chem Int Ed Engl. 1998;37:505–507. doi: 10.1002/(SICI)1521-3773(19980302)37:4<505::AID-ANIE505>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 25.Flom S R, Nagarajan V, Barbara P F. J Phys Chem. 1986;90:2085–2092. [Google Scholar]
- 26.Arai T, Karatsu T, Sakaraji H, Tokumaru K, Tamai N, Yamazaki I. Chem Phys Lett. 1989;158:429–434. [Google Scholar]
- 27.Mazzucato U, Momicchioli F. EPA Lett. 1992. March, 31–39. [Google Scholar]
- 28.Leigh W J, Postigo J A. J. Chem. Soc. Chem. Commun. 1993. 1836–1837. [Google Scholar]
- 29.Olivucci M, Bernardi F, Celani P, Ragazos I, Robbs M A. J Am Chem Soc. 1994;116:1077–1085. [Google Scholar]
- 30.Geravelli M, Celani P, Yamamoto N, Bernardi F, Robbs M A, Olivucci M. J Am Chem Soc. 1996;118:11656–11657. [Google Scholar]
- 31.Ishiguro M. J Am Chem Soc. 2000;122:444–451. [Google Scholar]
- 32.Saltiel J, D'Agostino J, Megarity E D, Metts L, Neuberger K R, Wrighton M, Zafiriu O C. Org Photochem. 1973;3:40–42. [Google Scholar]
- 33.Dellinger B, Kasha M. Chem Phys Lett. 1975;36:410–415. [Google Scholar]
- 34.Dellinger B, Kasha M. Chem Phys Lett. 1976;38:9–14. [Google Scholar]
- 35.Andersson P O, Gillbro T, Asato A E, Liu R S H. Chem Phys Lett. 1995;235:76–82. [Google Scholar]
- 36.Hammond G S. Adv Photochem. 1969;7:373–391. [Google Scholar]
- 37.Ramamurthy V, editor. Photochemistry in Organized and Constrained Media. New York: VCH; 1991. [Google Scholar]
- 38.Weiss R G, Ramamurthy V, Hammond G S. Acc Chem Res. 1993;26:530–536. [Google Scholar]
- 39.Dartnall H J A. Vision Res. 1967;8:339–358. doi: 10.1016/0042-6989(68)90104-1. [DOI] [PubMed] [Google Scholar]
- 40.Liu R S H, Shichida Y. In: Handbook of Photochemistry and Photobiology. Horspool W M, Song P-S, editors. Boca Raton, FL: CRC; 1995. pp. 817–840. [Google Scholar]
- 41.Lou J, Hashimoto M, Berova N, Nakanishi K. Org Lett. 1999;1:51–54. doi: 10.1021/ol990048l. [DOI] [PubMed] [Google Scholar]
- 42.Liu R S H, Asato A E, Denny M. J Am Chem Soc. 1983;105:2923–2924. [Google Scholar]
- 43.Turro N J., Jr . Modern Molecular Photochemistry. Inc., Menlo Park, CA: Benjamin/Cummings Publishing Co.; 1978. pp. 72–74. [Google Scholar]
- 44.Liu R S H, Asato A E. Tetrahedron. 1984;40:1931–1969. [Google Scholar]
- 45.Fischer E. J Photochem. 1981;17:331–340. [Google Scholar]
- 46.Castel N, Fischer E. J Mol Struct. 1985;127:159–166. [Google Scholar]
- 47.Alfimov M V, Razumov V F, Rachinsky A G, Listvan V N, Scheck Y B. Chem Phys Lett. 1983;101:593–597. [Google Scholar]
- 48.Yoshizawa T, Kandori H. Prog Retinal Res. 1991;11:33–55. [Google Scholar]
- 49.Asato A E, Denny M, Liu R S H. J Am Chem Soc. 1986;108:5032–5033. [Google Scholar]
- 50.Liu R S H, Browne D T. Acc Chem Res. 1986;19:42–48. [Google Scholar]
- 51.Sheves M, Albeck A, Ottolenghi M, Bovee-Geurts P H M, De Grip W J, Einterz C M, Lewis J M, Schaechter L E, Kliger D S. J Am Chem Soc. 1986;108:6440–6441. [Google Scholar]
- 52.Schulten K. In: Energetics and Structures of Halophilic Microorganisms. Caplan S R, Ginsberg M, editors. Amsterdam: Elsevier; 1978. pp. 331–334. [Google Scholar]
- 53.Liu R S H, Mead D, Asato A E. J Am Chem Soc. 1985;107:6609–6614. [Google Scholar]
- 54.Schulten K, Humphrey W, Logumov I, Sheves M, Xu D. Isr J Chem. 1995;35:447–464. [Google Scholar]
- 55.Mathies, R. A., Lin, S. W., Ames, J. B. & Pollard, W. T. (1991) Annu. Rev. Biophys. Chem.491–518. [DOI] [PubMed]
- 56.Genick U K, Soltis S M, Kuhn P, Canenestrelli I L, Getzoff E D. Nature (London) 1998;392:206–209. doi: 10.1038/32462. [DOI] [PubMed] [Google Scholar]
- 57.Hellingwerf K J. J Photochem Photobiol B. 2000;54:94–102. doi: 10.1016/s1011-1344(00)00004-x. [DOI] [PubMed] [Google Scholar]
- 58.Williams, R. E. & Braslavsky, S. E. (2000) Encyclopedia on Photobiology, in press.