

Abstract
A new Monte Carlo algorithm is presented for the efficient sampling of the Boltzmann distribution of configurations of systems with rough energy landscapes. The method is based on the introduction of a fictitious coordinate y so that the dimensionality of the system is increased by one. This augmented system has a potential surface and a temperature that is made to depend on the new coordinate y in such a way that for a small strip of the y space, called the “normal region,” the temperature is set equal to the temperature desired and the potential is the original rough energy potential. To enhance barrier crossing outside the “normal region,” the energy barriers are reduced by truncation (with preservation of the potential minima) and the temperature is made to increase with |y|. The method, called catalytic tempering or CAT, is found to greatly improve the rate of convergence of Monte Carlo sampling in model systems and to eliminate the quasi-ergodic behavior often found in the sampling of rough energy landscapes.
Keywords: simulated tempering | simulated annealing | enhanced sampling techniques | rough energy landscapes
In the Monte Carlo sampling of the conformation space of complex systems, one often encounters the problem of quasi-ergodicity. This problem arises from the roughness of the potential energy landscape where local energy minima are separated by large energy barriers. In such systems, a trajectory arising in one basin of attraction only infrequently crosses neighboring high energy barriers to explore a new thermally accessible basin. Although theoretically ergodic, the ordinary Metropolis algorithm fails to sample barrier crossings frequently enough to explore the accessible configuration space and thus evinces practical quasi-ergodicity. In proteins, for example, the energy barriers arise from at least two classes of interactions: first, local barriers separate stable torsion angle states; second, barriers arise from strong repulsive interactions between neighboring atoms on different side-chains. These strong repulsive forces lead to frustration when the side-chains become densely packed.
Many methods have been proposed to enhance the conformation space sampling (1–17). These methods include multicanonical sampling, simulated tempering, parallel tempering, and the method of expanded ensembles, including strategies based on increased dimensionalization.
Enzymes accelerate reactions in small molecular substrates by reducing the free energy barriers separating reactants from products without changing the relative positions of the potential minima. Can we use this strategy to accelerate the sampling of configuration space in simulations of complex systems? We propose a new method based on this idea which we call catalytic tempering or CAT. CAT is based in part on a barrier reducing strategy that does not change the location or values of the potential minima (4), thereby accelerating barrier crossing while keeping the equilibrium properties of the system unchanged, as in the action of a catalyst or enzyme.
In the CAT method, the dimensionality of the system is increased by the introduction of a fictitious coordinate y. This augmented system is given a potential energy function,
 |
1 |
where x designates the coordinates of the protein or other systems of interest, y is the new dimension whose domain is −ymax ≤ y ≤ ymax, and y is said to be in the normal region (n.r.) when −y0 ≤ y ≤ y0 for y0 ≤ ymax. The function h(y) is unity when y is in the n.r. and goes to zero (either smoothly or discontinuously) outside this region. The region outside the normal region will henceforth be called the “catalytic region” (c.r.). U(x) is the potential energy surface for the system to be sampled and UT(x) is the potential energy function with truncated barriers that we call the catalytic potential landscape. The constant V0 modifies the zero of both U and UT in a similar fashion. In Eq. 1, the function ξ(y) is taken to be ξ(y) = 1 if y is in the n.r. and ξ(y) is a decreasing function of |y| if y in the c.r. ξ(y) can be (but need not be) interpreted as the ratio of two temperatures, ξ(y) = T0/T(y), where the temperature T(y) is set equal to the desired temperature T0 in the n.r. but is allowed to increase with y in the catalytic region. In the simulations to be presented later, we take ξ(y) to be:
 |
2 |
Monte Carlo methods can then be used to sample the joint distribution function of x and y in this higher dimensional space,
 |
3 |
where Z(y; V0) = ∫ dx exp {−β0V (x, y; V0)}, β0 = 1/kBT0, and Q(y) is the normalized distribution function of the y-coordinate, which can be chosen arbitrarily. Notice that the term containing V0 cancels in Eq. 3. In the n.r. this distribution reduces to αQ(y) exp {−β0U(x)}, where α is a proportionality constant. Monte Carlo sampling of the joint distribution function PQ (x, y; V0) in the n.r. can thus be used to determine the desired distribution function
 |
4 |
It should be noted that V (x, y; V0) was defined so that in the catalytic region c.r. the temperature can be interpreted as an increasing function of |y| and the energy barriers are reduced by truncation. It is expected that CAT Monte Carlo sampling should lead to very efficient barrier crossings when y lies in the c.r. When y returns to the n.r., the system will be on the correct potential surface whose zero is calibrated with V0. The constant V0 can be used to control the sampling distribution of y as will be discussed in Methods. If we eliminate the catalysis by taking UT(x) = U(x) in Eq. 1 our method becomes similar to simulated tempering (10), with a continuous temperature and with a more general weighting. Introduction of the catalytic potential surface leads to considerable improvements over simulated tempering, greatly improves the rate of convergence of Monte Carlo sampling and eliminates quasi-ergodic behavior often found in the sampling of rough energy landscapes.
This paper is organized as follows. The method is described in the next section and is followed by applications to illustrative problems. The first system studied is that of a single particle moving on a one-dimensional random potential constructed from Gaussians. This represents a frustrated system and helps to clearly show how CAT is to be optimized. The second system is a one-dimensional Lennard–Jones mixture consisting of eight equal-length rods confined to a one-dimensional box and interacting through Lennard–Jones potentials with different well depths for each pair of particles. The repulsive core of each Lennard–Jones potential is softened by cutting it off at energies large compared with the thermal energy to make the system metrically indecomposable. The activation energy for two particles to pass through each other is now large but finite as oppposed to being infinite for the Lennard–Jones potential. The multidimensional potential energy surface will consist of basins of attraction corresponding to different rod orderings separated by large barriers with each basin consisting of wells separated by smaller barriers. This system will behave like a glass. Starting in one basin, Monte Carlo sampling will be quasi-ergodic so that at low temperatures one will not be able to move from one basin to another and land in the lowest energy basin let alone the lowest energy well within the lowest energy basin. We show that CAT very effectively samples this landscape and finds the lowest energies in the system.
Methods
Simply stated, CAT is a method to sample the Boltzmann distribution corresponding to the potential U(x) at temperature T0. Its implementation requires the specification of a number of parameters and functions, described next.
To implement CAT, we need the following ingredients: (i) a truncated version UT(x) of U(x); (ii) a maximum value ymax for the fictitious coordinate y; (iii) a range [−y0, y0] to define the n.r. in the y space; (iv) a choice of the function ξ(y). If we interpret ξ(y) = T0/T(y), then the choice of ξ reduces to the choice of temperature, which may be more intuitive. T(y) is chosen such that T(y) = T0 in the n.r., and increasing from T0 up to a chosen maximum value T(ymax) = Tmax in the c.r. (v) A choice of the switching function h(y). Here we choose it to be 1 in the n.r. and 0 in the c.r. (vi) A choice of the y-coordinate distribution Q(y), with which the y space is going to be sampled. (vii) The specification of the step sizes Δx and Δy for the Monte Carlo moves in the x and y directions, respectively.
With the above prescriptions, we apply the standard Metropolis algorithm to sample the distribution of Eq. 3. We implement our Monte Carlo scheme with two different kinds of local moves:
Moves xi → x′i in the configuration space, with uniform transition probability within the interval [xi − Δx/2, xi + Δx/2], and acceptance ratio
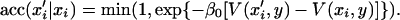 |
5 |
Moves y → y′ with uniform transition probabilities in [y − Δy/2, y + Δy/2], and acceptance ratio
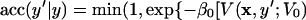 |
6 |
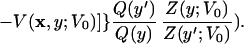 |
In the description of the Monte Carlo moves above, we must determine Z(y;V0) to within a constant (see Eq. 6). We now describe an iterative procedure for accomplishing this.
Assume we have the nth approximation Zn(y;V0) to Z(y;V0). A run using Zn instead of the exact Z in Eq. 6 would yield a y-probability density function that we shall call Qn+1 (y). On the other hand, integration of Eq. 3 over x yields that Z(y;V0) = Zn(y; V0)Qn+1(y)/Q(y). Given that the run from which Qn+1 was computed was finite, the previous relation can only be an approximation: the n+ 1 approximation Zn+1 to Z. Thus we obtain the recursive relation.¶
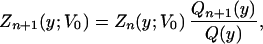 |
7 |
Assuming a limit exists, this relation converges (up to fluctuations due to the finiteness of the run) to the theoretical Z(y;V0). Along with Zn → Z, we also have that Qn → Q. That is the y distribution ends up being the one we chose. This allows us to schedule the protocol of tempering and catalysis.
When the recursion of Eq. 7 is started using Z0(y;V0) = Z(V0) Q(y) (where Z(V0) is the overall partition function, but here it only plays the role of a proportionality constant), the first iteration yields a y-probability density which is proportional to Z1(y;V0): Q1(y) = Z1(y;V0)/Z(V0). To make its relationship with the partition function explicit, we shall denote Q1 by QZ. Therefore,
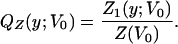 |
8 |
In some cases, for example for small systems, the first iteration is all that will be necessary to achieve convergence. Often, however, the first iteration will generate a distribution function QZ(y;V0) for which certain regions in y-space will have a very small probability. Then transitions across these regions will be very rare and CAT will break down. It will then be necessary to make many iterations to get convergence to the desired Q(y). There are many ways to implement this strategy. One way to generate the first iterate above is to determine that value of V0 that will make QZ(y;V0) as uniform as possible. This can be achieved by finding the V0 that maximizes the entropy of QZ(y;V0), following the method prescribed in the Appendix. This special value of V0 will be denoted by V*0 hereafter. The QZ(y;V*0) thus determined can then be used as the starting point for the next iteration. In the following, we test the adequacy of using the first iterate determined in this way to sample several simple systems with rough energy landscapes, leaving for a more extensive paper the application of the full iterative scheme to the problem of protein folding.
Let us recapitulate. The reason to find the optimal V*0 was to sample the y-coordinate as homogeneously as possible. Thus we can trust that the resulting numerical QZ(y;V*0) is close to the one given in 8, and solve for the partition function Z(y;V*0) = QZ(y;V*0)Z(V*0). Using this, the acceptance probability can be computed from Eq. 6 as
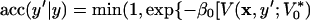 |
9 |
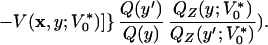 |
Our CAT scheme would be very inefficient if it were to make our system spend most of the time in the n.r., i.e., if Q(y) had most of its mass in the n.r. In such an event, no catalysis or tempering would take place. Likewise, if Q(y) had most of its mass outside of the n.r., then we would only rarely probe the potential U(x) that we actually wish to sample. We can choose Q(y) arbitrarily very much as we can choose the annealing schedule in an implementation of simulated annealing. Which Q(y) is “the best” is very much problem dependent. In particular implementations, one may wish to test various alternatives and choose the Q(y) that gives the best results. In this paper, the CAT scheme will be implemented with Q(y) = QZ(y;V*0).
In the original papers of Marinari and Parisi (10) and Lyubartsev et al. (13) (who discovered ST independently calling it the method of extended ensembles), as well as in other studies using ST (12), the temperature distribution is taken to be uniform. The optimal temperature distribution is probably problem dependent and thus should be chosen by trial and error on a case-by-case basis. It should be noted that if we wish to impose a particular temperature distribution P(T), we must choose Q(y) = P(T(y))|T′(y)|. If we chose to have a uniform temperature distribution, we could then choose T(y) to be a linear function of y and the resulting Q(y) will then be uniform as well.
In the subsequent sections, when we compare CAT with ST, we use two implementations of ST. In one of them, called UST, the temperature distribution is made uniform [as in the original formulations of ST (10, 13)] by taking a uniform Q(y) and a linear dependence of T(y) with y. We also explore a nonuniform temperature distribution by taking T(y) to be a linear function of y and a nonuniform Q(y) given by Q(y) = ZST(y;V0)/∫ dy ZST(y;V0), where ZST(y;V0) = ∫ dx exp {−β0VST(x, y;V0)} and VST(x, y;V0) = [U(x) − V0]ξ(y) is the potential used for ST. We shall refer to this implementation of ST as ZST.
Sampling a Random Potential
In this section, we consider the following one-dimensional potential:
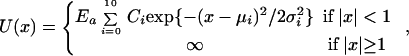 |
[10]
where the Ci were chosen randomly between 0 and 1, the μi lay on an equispaced grid whose lattice size is 1/6, and the σi were chosen randomly between 0 and 0.2. The resulting potential for Ea = 50 can be seen in the inset of Fig. 1 (solid line). Truncated versions of this potential were chosen by replacing the Gaussian functions by min (κ, exp {−(x − μi)2/2σi2}) (dashed lines in the inset of Fig. 1), where κ ≤ 1 is the truncation parameter.
Figure 1.

The decay of the square deviation given by Eq. 11 as a function of the number of Monte Carlo (MC) steps for the one-dimensional random potential of Eq. 10 and different Monte Carlo schemes (CAT, ZST, UST and classical Monte Carlo). The random potential is plotted in the inset for various values of κ which controls the level of truncation of the barriers. The other parameters of the Monte Carlo run are detailed above the figure. For the most severe truncation, CAT samples this potential 100 times faster than the simulated tempering methods.
The reason for studying the potential given in Eq. 10 is that it is a one-dimensional approximation of the energy landscape of glassy systems with barriers of different heights, where some of the barriers are high enough relative to the thermal energy to create quasi-ergodicity. The one-dimensional nature of this potential allows for an easy comparison between the numeric distribution being sampled with ST (both ZST and UST), CAT or regular Monte Carlo, and the theoretical one. For any of these sampling techniques, the numerical distribution corresponding to initial condition i, after t Monte Carlo steps shall be denoted by ρnum(x; t, i). The exact distribution, is obviously ρtheo(x) = Z−1 exp{−U(x)/kBT0}. If we have N initial conditions (in the simulation to be shown below N = 10), the quantity (1)
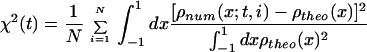 |
11 |
is a normalized global measure of the degree to which the numerical simulation approximates the exact distribution after a time t. In Fig. 1 we have plotted χ2 for classical Monte Carlo (open circles), for ZST (squares), for UST (diamonds), and for CAT (filled circles). We can see that classical Monte Carlo sampling is unable to sample the potential even after a billion iterations. This means that each of the 10 initial conditions (chosen randomly between [−1, 1]) got trapped in a well, and may be visiting other wells, but not fast enough to get a good sampling of the Boltzmann distribution even after this many iterations. The contrast with any of the other techniques, ZST, UST, and CAT, is remarkable. After a short run of 108 moves, both UST and ZST sampled the Boltzmann distribution with a relative error of less than 1%. ZST performs marginally better than UST. CAT, on the other hand, samples the desired distribution with a relative error of 1% in about 4 × 106 Monte Carlo steps for the truncation parameter κ = 0.5, and 106 Monte Carlo steps for κ = 0.25, i.e., 25 to 100 times faster than the ST schemes. This can mean the difference between a simulation taking an hour or several days!
Linear Array of Lennard–Jones Rods
In this section, we apply CAT to the problem of sampling an infinitely frustrated system of rods of length σ confined to a one-dimensional box of length L = 2. We assign energy parameters ɛi to each rod i so that the Lennard–Jones interaction between rods i and j is
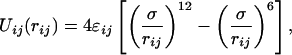 |
12 |
where we take ɛij =
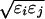 . Each ordering of the
particles will have an associated energy basin with several maxima and
minima. The (degenerate) global minimum energy configuration of the
system will correspond to specific orderings of the particles. Neither
ordinary Monte Carlo nor ST starting from a given ordering will ever be
able to sample any other ordering because none of the rods can pass
through each other in these methods. Of course, this is an artifact of
the Lennard–Jones potential and the one-dimensional nature of the
system. However, this system is a good “straw man” to show where
the strengths of CAT lay. CAT, on the other hand, should be able to
sample the minimum energy configuration because it can generate paths
that can pass over the truncated barriers. Thus we expect CAT to
generate an efficient sampling of the thermal distribution. We shall
see that such is the case.
. Each ordering of the
particles will have an associated energy basin with several maxima and
minima. The (degenerate) global minimum energy configuration of the
system will correspond to specific orderings of the particles. Neither
ordinary Monte Carlo nor ST starting from a given ordering will ever be
able to sample any other ordering because none of the rods can pass
through each other in these methods. Of course, this is an artifact of
the Lennard–Jones potential and the one-dimensional nature of the
system. However, this system is a good “straw man” to show where
the strengths of CAT lay. CAT, on the other hand, should be able to
sample the minimum energy configuration because it can generate paths
that can pass over the truncated barriers. Thus we expect CAT to
generate an efficient sampling of the thermal distribution. We shall
see that such is the case.
For simplicity, we study a system of N = 8 particles and assign particles labeled 1, 2, 3, and 4 the same value ɛS = 0.1ɛ and particles labeled 5, 6, 7, and 8 the value ɛL = ɛ. We call the first set of particles “small particles” (S) and the second set “large particles” (L) even though all of the particles have the same Lennard–Jones σ-parameter, that is, they have the same lengths. For this simple system we have 6 (4 choose 2) symmetric configurations (LLSSSSLL, LSLSSLSL, SLLSSLLS, LSSLLSSL, SLSLLSLS, and SSLLLLSS), each contributing its own basin energy minimum. Each of these energy minima has degeneracy 4!×4! = 576. With the remaining 64 (8 choose 4 minus 4 choose 2) assymetric configurations we can form pairs, by grouping the mirror image arrangements (e.g., LSSSSLLL and LLLSSSSL, etc.). It is clear that mirror image configurations will share the same energy minimum. Thus there will be 32 distinct energies corresponding to the assymetric pairs, and each of these energies has degeneracy 2×4!×4! = 1,152. The ordering corresponding to the global minimum energy is SSLLLLSS. Because this is a symmetric arrangement, there are 576 degenerate particle orderings corresponding to this global energy minimum which should be found among the 8! = 40,320 possible permutations. How well does CAT do?
In applying CAT we take for U(x) in Eq. 1 the full Lennard–Jones potential U(x1, … , xN) = Σi<j Uij(rij) where Uij(rij) is Lennard–Jones potential given in Eq. 12. We define the truncated Lennard–Jones potential to be
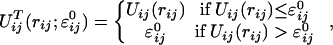 |
13 |
where ɛij0 is a parameter that defines the finite energy barrier. For N particles the total truncated potential to be used in Eq. 1 is UT(x1, … , xN) = Σi<j UijT (rij). Note that this truncation is making an infinite energy barrier become finite.
The simulations to be discussed below were performed using the following parameters: kBT0 = 1, ɛ = 5kBT0, ɛS = 0.1 ɛ and ɛL = ɛ. The function ξ was chosen as in Eq. 2, with kBTmax = 10 and ymax = 1. The length σ of the rods was taken to be 1/32, which gives sufficient free volume to the rods within the box of size 2. The scale of the local y-moves is Δy = 0.1, and the scale of the local x-moves is Δx = σ/2. Because of the considerable free volume, the acceptance probability for these Δx is around 90%. The energy at which the Lennard–Jones potentials are truncated (see Eq. 13) is taken as ɛij0 = ɛij.
As mentioned earlier, the point in the eight-dimensional space that represents the configuration of this system could not possibly visit different particle orderings in either the ordinary Monte Carlo or ST scheme. The different orderings in this eight-dimensional space form closed compartments, separated by infinite energy barriers. The state space is then metrically decomposable and the Monte Carlo will be strictly non-ergodic. Each of the decomposed subregions will have an energy surface characterized be many wells and barriers due to the attractive interactions between the particles. We call these subregions corresponding to each particle ordering an “energy basin.” This system can be made quasi-ergodic if one transforms the energy surface by cutting off the infinite barriers into finite but large barriers (compared to the thermal energy). In this way, the energy surface will be characterized by sizeable basins of attraction corresponding to different particle orderings separated by finite-albeit-large energy barriers. Ordinary Monte Carlo sampling on this surface for temperatures small compared to these large barriers will now be quasi-ergodic. The system will behave like a glass with similar structural frustration. This system has many of the characteristics of a “one-dimensional protein.” CAT adds one more coordinate to the system, and in the c.r. of this nine-dimensional space, the basins (strict compartments in the case of the infinite barriers) will be connected by much smaller barriers (instead of infinite or very high barriers) in the catalytic region. Thus a way to measure how well CAT deconfines the configuration-space point and facilitates its wandering in the configuration space, is to plot the number of distinct new orderings in the n.r. as a function of the number of iterations.
The potential we will be considering below is the Lennard–Jones potential whose repulsive region sampled by both CAT and ST is cut off at 50 ɛij to create the quasi-ergodic behavior. In CAT we have in addition truncated this potential at ɛij0 = 1.0 ɛij in the c.r. For ST we have made no additional truncation in this region. Fig. 2a shows the number of different configurations that the system has adopted vs. the number of iterations, where each configuration counted is in the n.r. for CAT (where V0 = −2.4 has been optimized by the maximum entropy technique, see Appendix). This is compared with ST by turning off the truncation of the potential. We observe for CAT a linear increase in the number of distinct configurations visited, with slightly over 1,600 configurations visited in total (solid circles) after a million iterations. The linear regime should eventually saturate, given that there are a finite number of possible configurations (40,320), but the asymptotic regime has not been reached yet. By comparison, ZST only generates four configurations (open squares in Fig. 2a) in an equivalent time and is thus more than two orders of magnitude less efficient than CAT. Obviously if we had not softened the infinite repulsion of the Lennard–Jones potential in the n.r., ST would never have generated a particle ordering different from its initial ordering but CAT would. In addition, we could have made CAT even more efficient by truncating at lower repulsive energies in the c.r.
Figure 2.

(a) Time course of the number of distinct rod-orderings in the linear array of Lennard–Jones rods for CAT (solid circles) and ZST (open squares). We used a truncated Lennard–Jones potential at 50ɛ, and for CAT further truncated in the c.r. at ɛ (see Inset). ZST visited only four different orderings after 106 iterations. For CAT the different orderings grows linearly with time. (b) Probability density functions of the energy surface for the linear array of Lennard–Jones rods in two independent runs of CAT (solid line and dotted line) and two independent runs of ZST (dashed line and dot dashed lines), after 106 Monte Carlo passes. The two distributions from the CAT runs are almost identical. The two distributions from the ZST are quite different. Energy in units of T0.
As discussed above, CAT provides a means to sample different configurations quickly. How well does it generate the energy distribution? In Fig. 2b we plot the probability density function (PDF) of the energies visited by the linear array of Lennard–Jones rods in two independent runs of CAT (solid line and dotted line) and two independent runs of ZST (dashed line and dot dashed lines). In each of these runs, the two initial configurations of the rods was chosen randomly and are distinctly different. The four runs are the result of 1 million Monte Carlo passes. Only 5–10% of these steps (those for which y is in the n.r.) contribute to the PDF. The two distributions from the CAT runs are almost identical, suggesting that the sampling of the energy surface at this temperature has converged. The two distributions from the ZST are quite different. Each of them covered only a portion of the energy values covered by the CAT-sampled PDFs. This clearly demonstrates that there are conformations yet unvisited by ZST that have been sampled by CAT.
There are conspicuous modes in the PDFs of Fig. 2b. These peaks correspond to different conformations of the system of rods. For example, the peak at −10kBT0 corresponds to conformations in which three large rods form a cluster, which is not interacting with the fourth large rod. There is a combinatorial number of different conformations even within the same basin, with several conformations corresponding to the same energy. An analysis of the modes of the PDFs of Fig. 2b reveals that the CAT runs have sampled the energies of all the possible conformations. That is not the case for the ZST runs. One of them (dot-dashed line) never left the original ordering. The other (dashed line) visited four different orderings, not including the one corresponding to the former run. The conformations accessible to the rods in each of these ZST runs was characteristic of the few visited basins, thus producing a very biased sampling of the energy landscape.
Conclusions
In this paper, we introduced CAT, a new Monte Carlo algorithm for the efficient sampling of the configuration space of systems with rough energy landscapes. The method is based on the introduction of a fictitious coordinate y so that the dimensionality of the system is increased by one. Monte Carlo sampling in this higher dimensional space leads to very efficient barrier crossings and eliminates the quasi-ergodicity of ordinary Monte Carlo as well as ST, and multicanonical methods for sampling rough energy landscapes. Although our method is implemented using the Metropolis Monte Carlo algorithm, it could equally well have been based on the hybrid Monte Carlo protocol or other stochastic dynamics protocols. When applied to the very glassy one-dimensional heterogeneous system of Lennard–Jones rods moving in one dimension, CAT was able to find both the lowest energy conformation and the correct energy distribution where simulated annealing, simulated tempering, parallel tempering and multicanonical Monte Carlo are destined to fail.
Also as with other methods, CAT can not solve the NP-Complete problem that arises from the exponential growth of local energy minima with the size of the system. Nevertheless, CAT should generate new configurations more efficiently than other methods. This follows from the fact that such methods as simulated tempering, are subcases of CAT. The introduction of the barrier reducing strategy is expected to lead to improvements over these other methods.
It is a simple matter to combine CAT with other methods for accelerating rapid barrier crossings on rough energy landscapes such as quantum annealing. Moreover, one can easily apply this to the force fields used in protein folding. Since those force fields contain nonbonding forces such as the Lennard–Jones potential between centers as well as torsion angle potentials, it is a simple matter to design truncated potentials for CAT much as we have been able to do here. We are currently initiating a study on realistic proteins. We believe that CAT will allow us to sample the protein efficiently.
Although we have presented one method for finding a catalytic surface (i.e., a truncated potential) in which we have truncated each pairwise repulsive interaction, it should be clear that there are many other strategies that could be adopted instead. For example, it may be possible to generate an efficient CAT by truncating the whole potential energy surface uniformly at a given energy.
In closing, it is important to state that CAT can also be used as a method for annealing. One can sample states at reasonably low temperatures. Energy minimization from such sampled states should then determine the global energy minimum. It is also a simple matter to use CAT as a tool for generalizing simulated annealing. We call this method catalytic annealing (CAN). Thus, we are eager to see if barrier reduction in sampling (or BRIS) of which CAT is one example will be as efficient for higher dimensional systems.
Acknowledgments
We thank M. O. Magnasco whose paper “Changing the pace of evolution” (18) influenced our initial thinking about this problem. We also thank B. D. Silverman, A. K. Royyuru, and Yonghan Lee for fruitful discussions, and John Straub for a critical reading of the manuscript.
Abbreviations
- CAT
catalytic tempering
- n.r.
normal region
- c.r.
catalytic region
- ST
simulated tempering
- UST
simulated tempering with a uniform temperature distribution
- ZST
simulated tempering with a non-uniform temperature distribution
Appendix
To choose the value of V0 that maximizes the entropy of QZ(y;V0) it is convenient to make use of the functional dependence of V(x, y;V0) on V0 to derive a simple formula that relates the QZat two different values of V0. In effect, it is easy to show from Eq. 1 that
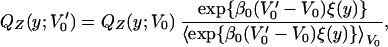 |
A1 |
where 〈f(y)〉V0 denotes an average of f(y) with respect to QZ(y;V0). Thus, if one has a sample of y corresponding to a given V0, we can calculate the entropy of the y-distribution corresponding for any V′0,
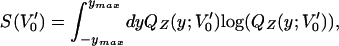 |
A2 |
and choose the value V*0 that maximizes this entropy. In this way, one can start with any value of V0, e.g., V0 ≡ V0(0) = 0 and sample PQZ(x,y;V0(0)) to obtain its marginal y distribution QZ. With the numerically obtained QZ(y;V0(0)), and using A1 to compute QZat other values of V′0, maximize A2 to compute the optimal V0(1). This precedure can be repeated starting with V0(1). This provides a recipe for iterating until the sequence V0(0) → V0(1) → V0(2) → ⋯ → V0(j) → ⋯ converges to the optimal V*0. In practice, this iteration can be stopped after a few cycles.
Footnotes
This iterative scheme is similar in spirit to one proposed in the literature (17) for simulated tempering. This equivalence can be understood if we define Zn(y) = exp(gn(y)). Then our iterative scheme is gn+1(y) = gn(y) + ln[Qn(y)/Q(y)].
References
- 1.Cao J, Berne B J. J Chem Phys. 1989;92:1980–1985. [Google Scholar]
- 2.Berg B A, Neuhaus T. Phys Lett B. 1991;267:249–253. [Google Scholar]
- 3.Swendsen R H, Wang J S. Phys Rev Lett. 1987;58:86–88. doi: 10.1103/PhysRevLett.58.86. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Berne B J. J Chem Phys. 1993;99:6071–6077. [Google Scholar]
- 5.Li Z, Scheraga H A. Proc Natl Acad Sci USA. 1987;84:6611–6615. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nayeem A, Vila J, Scheraga H A. J Comp Chem. 1991;12:594–605. [Google Scholar]
- 7.Guarnieri F, Still W C. J Comp Chem. 1994;11:1302–1310. [Google Scholar]
- 8.Senderowitz H, Guarneri F, Still W C. J Am Chem Soc. 1995;117:8211–8219. [Google Scholar]
- 9.Frantz D D, Freeman D L, Doll J D. J Chem Phys. 1990;93:2769–2784. [Google Scholar]
- 10.Marinari E, Parisi G. Europhys Lett. 1992;19:451–458. [Google Scholar]
- 11.Huksima K, Nemoto K. J Phys Soc Japan. 1996;65:1604–1608. [Google Scholar]
- 12.Hansmann U H E, Okamoto Y. Physica A. 1994;212:415–437. [Google Scholar]
- 13.Lyubarsev A P, Martsinovski A A, Shevkunov S V R, Vorontsov-Velyaminov P N. J Chem Phys. 1992;96:1776–1783. [Google Scholar]
- 14.Faken D B, Voter A F, Freeman D L, Doll J D. J Phys Chem. 1999;103:9521–9526. [Google Scholar]
- 15.Beutler T C, van Gunsteren W F. J Chem Phys. 1994;101:1417–1422. [Google Scholar]
- 16.Berne B J, Straub J E. Curr Topics Struct Biol. 1997;7:181–189. doi: 10.1016/s0959-440x(97)80023-1. [DOI] [PubMed] [Google Scholar]
- 17.Kerler W, Rehberg P. Phys Rev E. 1996;50:4220. doi: 10.1103/physreve.50.4220. [DOI] [PubMed] [Google Scholar]
- 18.Magnasco M O, Thaler D S. Phys Lett A. 1996;221:287–292. [Google Scholar]