

Abstract
Leukocyte adhesion under flow in the microvasculature is mediated by binding between cell surface receptors and complementary ligands expressed on the surface of the endothelium. Leukocytes adhere to endothelium in a two-step mechanism: rolling (primarily mediated by selectins) followed by firm adhesion (primarily mediated by integrins). Using a computational method called “Adhesive Dynamics,” we have simulated the adhesion of a cell to a surface in flow, and elucidated the relationship between receptor–ligand functional properties and the dynamics of adhesion. We express this relationship in a state diagram, a one-to-one map between the biophysical properties of adhesion molecules and various adhesive behaviors. Behaviors that are observed in simulations include firm adhesion, transient adhesion (rolling), and no adhesion. We varied the dissociative properties, association rate, bond elasticity, and shear rate and found that the unstressed dissociation rate, kro, and the bond interaction length, γ, are the most important molecular properties controlling the dynamics of adhesion. Experimental kro and γ values from the literature for molecules that are known to mediate rolling adhesion fall within the rolling region of the state diagram. We explain why L-selectin-mediated rolling, which has faster kro than other selectins, is accompanied by a smaller value for γ. We also show how changes in association rate, shear rate, and bond elasticity alter the dynamics of adhesion. The state diagram (which must be mapped for each receptor–ligand system) presents a concise and comprehensive means of understanding the relationship between bond functional properties and the dynamics of adhesion mediated by receptor–ligand bonds.
Trafficking of blood-borne cells into tissues is crucial to the proper function of the immune response. Inflammation, lymphocyte homing, and bone marrow replenishment after transplantation all depend on trafficking (1). Trafficking is mediated by receptor-mediated adhesion of blood-borne cells to the endothelial cells that line blood vessels. The transition from an unbound blood cell to an adherent one in flow involves a number of steps: initial tethering, transient “rolling” adhesion, and firm adhesion. Firm adhesion is usually followed by morphological changes and trans-endothelial migration into the tissue stroma, so that the cell can carry out its intended function within the tissue.
Different adhesion molecules mediate different stages in this multistep adhesion process. Transient rolling adhesion occurs when receptor–ligand bonds between the leukocyte and endothelium exert a friction on the leukocyte, such that its velocity drops well below the hydrodynamic velocity for an unencumbered leukocyte at the same separation distance and wall shear rate. In the cell biology literature, rolling is often defined as a significant decrease in velocity, to perhaps 50% or less of the hydrodynamic velocity for cells near a surface, VH (2). Rolling is mediated by a variety of adhesion molecules, including selectins (3–6) and two α4 integrins (7, 8). Many adhesion molecules [e.g., β2 integrins binding to intercellular adhesion molecule 1 (ICAM-1) (9)] can mediate adhesion in the absence of flow but are not able to mediate attachment in the presence of flow. This suggests that there is a “no adhesion” state for cell attachment in flow. In addition, many antigen–antibody pairs mediate “firm adhesion” under flow (10, 11), characterized by sudden arrest from free-stream motion followed by durable attachment. In inflammation, firm adhesion can be mediated by activated β2 integrins once the leukocytes have been slowed by selectin-mediated rolling (9, 12). α4 integrins can also mediate firm adhesion when activated (7, 8) and may, through conformational changes, mediate both “firm” and “transient” types of adhesion. A sampling of antigen/antibody pairs shows that some mediate transient adhesion, some mediate firm adhesion, and some do not mediate adhesion at all (13). Thus, different dynamic states of adhesion are mediated by different adhesion molecules. The question arises: what functional properties of these molecules control the different dynamics of adhesion?
Evidence that the dynamics of adhesion is coded by the physical chemistry of adhesion molecules, and not by cellular features such as deformability, morphology, or signaling, comes from our laboratory's cell-free adhesion experiments (6, 14). We attached the selectin-binding tetrasaccharide sialyl-Lewisx to inert, rigid polystyrene microspheres, and demonstrated that these model leukocytes roll over E-, P-, or L-selectin substrates at velocities comparable to those of similarly sized real leukocytes. These results suggest that adhesive behavior can be modulated by cell deformability, morphology, and signaling, but is determined primarily by the physical chemistry of adhesion molecules.
Possible physicochemical properties that give rise to the various dynamic states of adhesion are rates of reaction, affinity, mechanical elasticity, kinetic response to stress, and length of adhesion molecules. Intuitively, we expect the dissociation rate of the bond and its dependence on force to be important in rolling adhesion. In rolling, bonds are subjected to stress, particularly at the trailing edge of the contact zone. The acceleration of bond dissociation caused by applied stress will affect the cell's ability to sustain rolling adhesion, and the time scale for bond rupture will dictate the rolling velocity. Bell (15) proposed that the net dissociation rate, kr(f) of a bond under applied force, f, could be given by:
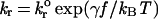 |
1 |
where kro is the unstressed dissociation rate constant, kBT is the thermal energy, and γ is a parameter with units of length that relates the reactivity of the molecule to the distance to the transition state in the intermolecular potential of mean force for single bonds (15, 16). The “Bell model” parameters kro and γ are functional properties of the molecules. Other models that describe the relationship between dissociation and stress exist (16, 17), yet the Bell model has been shown to be a good approximation for a sharp transition state (18, 19). Measurements of the Bell model parameters for adhesion molecules have been made using two independent methods: arrest duration distribution of cells on sparse coatings of adhesion molecules and dynamic force spectroscopy. The latter technique shows that the Bell model is valid, with distinct values of the model parameters for different regimes of force loading rate, rf. For leukocyte rolling, loading rates are >103 pN⋅s−1, a regime for which the use of a single set of Bell model parameters is plausible.
Dembo et al. (17) showed, using calculations of a membrane peeling from a surface, that dissociative properties are critical in determining whether and how fast cells would roll. Using Adhesive Dynamics, we can simulate the dynamics of cell attachment, rolling, and firm adhesion to a surface in flow (20). Using the Bell model for the force dependence of dissociation, we performed Adhesive Dynamics computer simulations to develop a “state diagram” for the states of adhesion that emerge for different values of the Bell model parameters. We also examined the influence of association rate, molecular elasticity, and wall shear stress on the state diagram. We show that adhesive behavior in flow is controlled primarily by the Bell model parameters. Using the simulation, one may thus predict the adhesive behavior mediated by various receptor–ligand pairs from the properties of single adhesion molecules measured by using arrest duration distribution in shear (21), force spectroscopy (22), or atomic force microscopy (23).
Methods
The Adhesive Dynamics method (shown schematically in Fig. 1) has been extensively described (20, 24, 25). The simulation begins with a freely moving “cell” or receptor-coated particle (modeled as a sphere with receptors distributed at random over its surface) at a separation distance, h, greater than the length of a receptor–ligand bond. The cell is allowed to reach a steady separation distance and translational velocity in the absence of specific interactions, after which receptor-mediated binding is initiated. The tip of each free adhesion molecule and the uniformly reactive substrate react with association rate kf and dissociation rate kr. We use Eq. 1 to model kr. To model the forward reaction, we note that kf is a function of both h and the slip velocity, Vs, between the cell and the surface (26). We assume that kf is equal to its maximum value kro(Vs), provided h is less than a critical separation distance, Hc, and kf = 0 for h > Hc. During each time step, bond formation and breakage are simulated by a Monte Carlo lottery, in which random numbers are compared with the probabilities for binding and unbinding to determine whether a bond will form or break in the time interval (24). The bond stresses, f, are calculated from the distance between the end points of the attachment, L, using Hooke's law, f = σ(L − λ), where σ is the spring constant. The stress contributed by each bond is summed to determine the total force and torque exerted by the bonds on the cell. In addition to the bonding forces, we include colloidal forces (24), and the external force and torque imparted to the cell by fluid shear (27) to compute the net force and torque acting on the cell. The motion of the particle is obtained from the mobility matrix for a sphere near a plane wall in a viscous fluid (20, 24). The new positions of free receptors and tethers at t + dt are updated from their positions at t, using the translational and angular velocity of the cell. The process is repeated until the cell travels 0.1 cm, or 10 s of simulated time has elapsed.
Figure 1.

Schematic diagram of Adhesive Dynamics. h is the separation distance between cell and surface. Receptors inside the region defined by Hc are reactive.
The simulations in this paper are performed at a constant wall shear rate of 100 s−1, unless otherwise noted. This shear rate is in the center of the range for which leukocyte rolling is observed (28, 29). The mean velocity and arrest duration can be obtained from the trajectory of position as function of time. The fractional stop time is the fraction of total time that the cell remains motionless (V < 0.01VH).
Results
Parameters used for the simulations are shown in Table 1. The particle size and shear rate are chosen to match the values from selectin mediated cell-free rolling experiments (6). The forward reaction rate, kf = 84 s−1, is a reasonable value that extensive simulation shows can properly recreate experimental values for velocity and dynamics of rolling.
Table 1.
Simulation parameters
| Parameter | Definition | Value (ref.) |
|---|---|---|
| Rc | Cell radius | 5.0 μm (14) |
| Rp | Receptor radius | 1.0 nm (15) |
| Nr | Receptor number | 25,000 (6) |
| NL | Ligand density | 3,600 cm−2 (6) |
| λ | Equilibrium bond length | 20 nm (15) |
| σ | Spring constant | 100 dyne⋅cm−1 (30) |
| μ | Viscosity | 0.01 g⋅cm−1⋅s−1 |
| G | Shear rate | 100 s−1 |
| Hc | Cut-off length for formation | 40 nm |
| T | Temperature | 310 K |
| kf | Association rate | 84.0 s−1 |
Adhesive Behavior States.
We find that there are at least four distinct, observable dynamic states of adhesion. Sample trajectories that demonstrate these behaviors are shown in Fig. 2. Fig. 2A shows the “no adhesion” state where cells are moving at a velocity greater than 50% of their hydrodynamic velocity, VH. “Fast adhesion,” in which cells move at V < 0.5VH but exhibit no durable arrests, is shown in Fig. 2B. Fig. 2C shows “transient adhesion” for which cells travel at V < 0.5VH but experience durable arrests. In “firm adhesion” (shown in Fig. 2D) cells bind and remain motionless. An “arrested” cell is defined as one that has moved <0.1 μm within 0.5 s, a criterion that can separate arrests from slow rolling, since 0.5 s is an easily accessible observation time in cell rolling assays (31). The “saltation” state, shown in Fig. 2E, is characterized by brief periods of firm arrest separated by periods of flow at VH. Leukocyte rolling falls within the transient adhesion regime, as it typically involves extended arrests and mean velocities below 0.5VH (6, 12, 31, 32). In the examples above, the reactive compliance parameter γ is held constant while kro is allowed to vary; similar differences in dynamic states may be observed by changing γ at constant kro (trajectories not shown). Thus, depending on the values of the Bell model parameters, all of the different adhesive behaviors may be observed.
Figure 2.

Representative trajectories of cell motions for all adhesive behaviors observed. Arrows indicate when interaction with the substrate begins. For A–D, γ = 0.01 Å; kf = 84 s−1. In E, γ = 0.001 Å; kf = 10 s−1. The kro values were A, 1,000 s−1; B, 200 s−1; C, 100 s−1; D, 20 s−1; and E, 10 s−1. The ratio of cell velocity to hydrodynamic velocity, V/VH, is A, 52%; B, 9%; C, 2%; D, 0%; and E, 3.4% (VH = 239 μm⋅s−1). In C, the motion is interrupted by an arrest of 4 s. In D, the cell is fully adherent (V = 0). In E, the flat sections of the trajectory have an average velocity about 0.01VH, and the steep sections have velocities ranging from 0.5VH to 0.8VH.
“State Diagram” for Adhesion.
To determine the adhesive behavior expected for a given set of Bell model parameters, we have calculated a state diagram for adhesion (Fig. 3), in which observed adhesive behaviors are plotted for a range of several orders of magnitude in kro and γ. The curve separating the states of no adhesion and fast adhesion is parametrized by a mean velocity of 0.5VH. The interpolated constant-velocity curves corresponding to 0.3VH and 0.1VH are also shown. The two curves that border the transient adhesion domain are based on the fractional stop time (arrest time/total time). The upper curve bounding this state corresponds to a fractional stop time of 0.01. The lower curve corresponds to a fractional stop time of 0.7.
Figure 3.

The state diagram for adhesion. Four different states are labeled. The dotted curve represents velocity of 0.3VH and the dashed curve represents velocity of 0.1VH. The locations corresponding to the Bell model parameters from Table 2 for E-selectin [squares (33, 34)], P-selectin [triangles for wild type (21, 34, 35) and gray triangles for mutants (35)], L-selectin [diamonds for wild type (34–36) and gray diamonds for mutants (35)], and peripheral node addressin (PNAd) [filled downward-pointing triangles (33, 34)] lie almost entirely within the envelope for rolling. The only exception is the P-selectin/P-selectin glycoprotein ligand 1 (PSGL-1) value from atomic force microscopy [cross (23)]. The points in red are from the Springer lab, the points in black or gray are from the McEver lab (35), and the points in blue are from the Lawrence lab (34).
From the state diagram, both kro and γ must have tightly bounded values for fast or transient adhesion to be observed. Fast or transient adhesive behaviors appear only in a thin strip of the state diagram, with the widest section corresponding to a 20-fold change in kro. For small γ, the domains corresponding to these states are horizontal, indicating that kro controls the cell motion, because γ is too small to increase kr substantially from kro at this shear rate. At larger values of γ, the curves are no longer horizontal, indicating that changes in both kro and γ regulate cell motion in this regime. When γ > 0.2 Å, cells traveling at V < 0.3VH exhibit significant periods of arrest. As γ increases beyond 0.2 Å, the curve parametrizing 0.3VH falls entirely within the transient adhesion state. Thus, rolling in this regime would be quite noisy, interrupted frequently by arrests. To maintain rolling when γ is high, kro must be small, and thus the arrest times become longer. When γ > 1 Å, the no adhesion state dominates the state diagram (for selectins). This lack of adhesion suggests that when γ > 1 Å, stresses imparted to the bond at this wall shear stress are large enough to cause rapid dissociation, and thus adhesion cannot be maintained.
A comprehensive list of Bell model parameters that have been reported in the literature for a variety of adhesion molecules by using the arrest duration distribution and force spectroscopy methods is given in Table 2. The Bell model parameter pairs measured for E-, P-, and L-selectin are plotted in Fig. 3 (the other Bell model parameter pairs will be discussed below). As expected, the parameter values for the selectins and their ligands fall within the transient adhesion section of the state diagram. Because it is known from independent experiments that these molecules mediate rolling adhesion, their location suggests that the state diagram can self-consistently predict the dynamic state of adhesion from molecular properties. The only point that does not fit is the value obtained by Fritz et al. (23), using atomic force microscopy. It is possible that the reconstituted system used by Fritz et al. had a different valency or molecular elasticity from the values obtained from leukocyte arrest duration distributions (all of the other data points). Because, as shown below, the state diagram is generated for one value of valency or elasticity, the position of the rolling envelope changes somewhat as valency or elasticity are varied.
Table 2.
Bell model parameters
| Receptor–ligand pair (ref.) | γ, Å | kro, s−1 |
|---|---|---|
| E-selectin–neutrophil (33) | 0.31 | 0.7 |
| E-selectin–neutrophil (34) | 0.18 | 2.6 |
| P-selectin–neutrophil (21) | 0.40 | 0.93 |
| P-selectin–neutrophil (34) | 0.39 | 2.4 |
| P-selectin–PSGL-1 (23) | 2.5 | 0.022 |
| P-selectin–PSGL-1 (35) | 0.29 | 1.1 |
| P-selectin mutant–PSGL-1 (35) | 0.24 | 1.8 |
| P-selectin mutant–PSGL-1 (35) | 0.33 | 1.7 |
| P-selectin mutant–PSGL-1 (35) | 0.42 | 1.6 |
| L-selectin–neutrophil (36) | 0.24 | 7.0 |
| L-selectin–neutrophil (34) | 1.11 | 2.8 |
| L-selectin–PSGL-1 (35) | 0.16 | 8.6 |
| L-selectin mutant–PSGL-1 (35) | 0.15 | 12.7 |
| L-selectin mutant–PSGL-1 (35) | 0.12 | 17.3 |
| L-selectin mutant–PSGL-1 (35) | 0.11 | 18.3 |
| PNAd–neutrophil (33) | 0.20 | 6.8 |
| PNAd–neutrophil (34) | 0.59 | 3.8 |
| Streptavidin–biotin rf < 104 pN⋅s−1 (22) | 5.1 | 0.013 |
| Streptavidin–biotin rf > 104 pN⋅s−1 (22) | 1.0 | 26.9 |
| Protein A–IgG (37) | 7.3 | 0.12 |
| PM-81 antibody–neutrophil (13) | 0.88 | 2.0 |
Association Rate.
The kf value used for the simulations shown in Fig. 3 is sufficiently high to guarantee receptor–ligand binding and to ensure that the association reaction is transport-limited (26). The ligand concentration of 3,600 μm−2, and the net association rate constant for each receptor, kf = 84 s−1, were chosen to match the conditions in cell free rolling experiments (6, 14). Fig. 4 shows the dynamics of adhesion for kf = 1 s−1 and 10 s−1. For clarity, only the boundaries of the transient and fast rolling states (the 0.5VH curve and firm adhesion curve) are shown. The rolling envelope shifts to lower values of kro as kf decreases. Thus for fixed kro and γ, cell rolling velocity increases with decreasing kf. The area of the rolling envelope decreases as kf decreases, and the rolling envelope terminates at smaller values of γ, such that for larger γ rolling is simply not possible regardless of kro. These observations support the conclusion that changes in kf do not drastically change the shape of the state diagram; rather the location of the rolling envelope is shifted in the kro–γ plane.
Figure 4.

The boundaries of rolling states for three different association rates: kf = 1.0, 10, and 84 s−1. For each rolling state, the upper boundary represents the 0.5VH curve, whereas the lower curve represents the border of the firm adhesion state.
As kf decreases, the type of motion observed within the rolling envelope also changes. At kf = 10 s−1, the transient adhesion state disappears, and cells roll without stops. As kf decreases further, a new state of adhesion appears in which cells interact with a surface at a velocity that alternates between two different values, one close to VH, the other close to 0.01VH. A typical trajectory of a cell in this saltation state is shown in Fig. 2E.
Wall Shear Rate.
Thus far, calculations have been for one wall shear rate, G = 100 s−1. We expected that firmly adherent cells might roll at higher shear rates, thus we calculated the firm adhesion boundary for G = 400 s−1. In addition, noninteracting cells may roll at lower shear rates, so we calculated the no adhesion boundary for G = 30 s−1. Note that the shear rates are chosen to cover the physiological range for postcapillary venules (38, 39). The outer boundaries for firm adhesion at 400 s−1 and no adhesion at 30 s−1 are plotted in Fig. 5. The dotted lines show the no adhesion and firm adhesion boundaries at G = 100 s−1 from Fig. 3. As expected, these changes in shear rate expand the range of Bell model parameters that can support rolling for some part of the physiological range of shear rate. The area above the upper boundary represents cells that exhibit no adhesion (V > 0.5VH) for all shear rates within 30–400 s−1. The firm adhesion region represents cells that remain arrested when G = 400 s−1. For γ > 0.2 Å and small kro, we find a new regime—“bimodal adhesion,” where cells are nonadherent at the higher shear rate (400 s−1) even though they were firmly adherent at the lower shear rate (100 s−1). No state of transient adhesion is seen, however, as the shear rate is increased. As γ increases further, the no adhesion behavior dominates, indicating that cells with Bell parameters in this region cannot maintain any type of adhesion even when G = 30 s−1. The remaining portion in the middle of the diagram—rolling adhesion—represents parameter values that mediate cell rolling over some part of the range of shear rates between 30 and 400 s−1.
Figure 5.

The state diagram with shear rate ranging from 30 to 400 s−1. The dotted curves indicate the boundaries of the rolling state at shear rate = 100 s−1. The rolling adhesion area represents the region of parameter space where rolling motion occurs over some part of the shear rate range from 30 to 400 s−1. The no adhesion regime indicates that cell rolling velocity is always larger than 0.5VH even when G = 30 s−1. The firm adhesion zone indicates that cells remain motionless even when G = 400 s−1. In the bimodal adhesion regime cells display either firm adhesion or no adhesion, without displaying rolling, as the applied shear rate is altered from 100 s−1 to 400 s−1.
Although our main interest was in simulating shear rates under 400 s−1, we also examined how the firm adhesion regime is altered when shear rate is increased beyond 400 s−1 (not shown). We find that there is often an abrupt transition from firm interaction to no adhesion as shear rate is increased. This behavior is like that observed in the bimodal adhesion regime, in which there is an abrupt transition from firm adhesion to no adhesion as shear rate is increased, with no rolling motion observed in between.
Bond Elasticity.
Thus far, we have set the elasticity of the receptor–ligand bond, σ, to 100 dyne⋅cm−1 (1 dyne = 10 μN). To determine the effect of elasticity of receptor–ligand bonds on cell adhesion, we performed a set of simulations with spring constant σ = 200 dyne⋅cm−1. The boundaries of rolling states for two different spring constants are plotted in Fig. 6. The rolling regime for σ = 200 dyne⋅cm−1 bends downward more steeply at larger values of γ. This result indicates that cells with stiffer bonds will roll faster, and the dissociation rate must be lower to compensate for this effect. Bonds with a larger σ respond more quickly to external forces, which makes kr increase more rapidly. As a result, under the same stress, bonds with a larger σ are more likely to break first. For larger values of γ, the rolling regime for σ = 200 dyne⋅cm−1 is seen at lower values of kro.
Figure 6.

The boundaries of rolling states for two different spring constants: σ = 100 and 200 dyne⋅cm−1.
Discussion
This paper presents a comprehensive state diagram that is able to describe how different dynamic states of adhesion in flow derive from the physical chemistry of individual receptor–ligand bonds. We have used the Bell model for the force dependence of dissociation rate, although any model for this relationship could have been used (16, 17).
In our simulations, we used a virtual “cell”—a hard sphere uniformly coated with adhesive receptors. Real cells, in contrast, are deformable and rough, and can undergo changes in receptor density, cytoskeletal attachment, and membrane rigidity caused by signaling. These cellular features likely modulate the dynamics of adhesion in ways that we have not included. Our “virtual cell” approach, in which the physics are completely specified by the “experimenter,” demonstrates the sufficiency of the physical chemistry of the adhesion molecules and their ligands to explain rolling. Deformability and signaling modulate when and where adhesion occurs, but they do not determine the type of adhesive behavior that will be observed. Support for this view comes from both partially and totally cell-free systems that can mimic leukocyte rolling. Our lab has shown that rigid, inert polystyrene microspheres coated with carbohydrates such as sialyl-Lewisx roll on substrates coated with E-selectin in a flow chamber (6, 14). The Springer group has shown that formaldehyde-fixed cells can roll on selectin surface (3) and that leukocytes or selectin-expressing transfected cells can roll over carbohydrate substrates (40). These studies emphasize that the physical chemistry of the adhesion molecules is the single most important factor controlling the dynamics of adhesion under flow.
Remarkably, almost all of the currently available Bell model parameter values for selectins (molecules that are known to mediate leukocyte rolling) fall within the transient (rolling) adhesion envelope in the state diagram (Fig. 3). The Springer lab has performed arrest duration distribution studies with neutrophils tethering on P-selectin (21) (which binds to the glycoprotein PSGL-1 on neutrophils), E-selectin (33) [which likely binds to many different molecules that bear sialyl-Lewisx (41)], L-selectin (36) (binding to PSGL-1), and the L-selectin ligand PNAd (33). The McEver lab has obtained results for L-selectin and P-selectin by using native PSGL-1 as well as PSGL-1 mutants that lack sulfation sites (35). The Lawrence lab has repeated these experiments with a high-speed camera to enable them to see extremely short-lived adhesive events (34). It is remarkable that the experimental values of the Bell model parameters for the selectins cluster to such a tight region in the state diagram. It is likely that because they have similar carbohydrate binding chemistries they all have similar values of kf, and thus plotting them all together on the same state diagram (produced for kf = 84 s−1 and NL = 3,600 cm−2) is reasonable.
The only selectin data that do not fall in the transient adhesion envelope are those from Fritz et al. (23) and two of the L-selectin-related points from Smith et al. (34). Concern about the data of Fritz et al. is raised by the discrepancy between their BIAcore measurements of PSGL-1/P-selectin reaction rates and those from the McEver lab (42). There may also be issues of viability and valency, since they did not perform rolling assays to test whether the molecules they used could mediate rolling. The simulations do describe most of the data, but experimental inconsistencies need to be resolved through further experimentation.
If plotted on a state diagram for the selectin parameters from Table 1, virtually all of the non-selectin Bell model parameter values from Table 2 fall in the region where even at G = 30 s−1 they would not be expected to mediate adhesion in flow. These predictions are not correct. One antibody/carbohydrate pair (PM-81) that displays transient adhesion (13) falls within the no adhesion envelope. Similarly, firm adhesion behavior has been observed for adhesion in flow mediated by streptavidin–biotin binding, even though the Bell model parameters from force spectroscopy fall in the no adhesion region of the state diagram.
The solution to these apparent discrepancies is that the state diagram shown in Fig. 3 applies to the specific combination of kf and NL used in selectin-mediated cell-free rolling experiments. The PM-81 and streptavidin data need to be placed on diagrams that reflect the proper values of kf and NL values used in their experiments. For receptor–ligand systems with very different association rates or molecular elasticities, the lines on the state diagram that delimit the different adhesive states will likely be shifted enough that firm adhesion would be the expected behavior. As Fig. 4 indicates, an increase in kf will shift transient adhesion envelopes to the right in the kro–γ plane, making any given value of kro–γ more adhesive. This observation highlights the important caveat that there are actually a family of state diagrams for which the shapes of the envelopes for rolling and firm adhesion are similar, but may shift depending on the value of the association rate, substrate ligand density, number of receptors on the cells, particle size, and the exact experimental protocol.
To address the issue of envelope shift, we have examined how kf, shear rate, and elasticity alter adhesive behavior. Association rate and shear rate control the adhesion in predictable ways: higher association rates and lower shear rates expand the adhesion envelope. The elasticity used in our simulations is σ = 100 dyne⋅cm−1, which is an average value for proteins (30). The Young modulus for proteins can vary greatly, however, from 1011 dyne⋅cm−2 (α-helices) to 107 dyne⋅cm−2 (elastin) (43, 44), and the exact value of the spring constant for the bonds that mediate leukocyte rolling is not known. Increasing the elasticity leads to a decrease in adhesiveness, a surprising and counterintuitive result, deriving from the ease with which stiff bonds can support a larger load at smaller deflections, which leads to a faster increase in dissociation under a fixed applied load.
The curved shape of the envelope for rolling explains why L-selectin has a higher kro and lower γ value than E- or P-selectin. The state diagram predicts that a higher value of kro must be accompanied by a lower value of γ to maintain rolling. This behavior has been reported by Alon et al. (33) for L-selectin. The higher unstressed off rate of L-selectin bonds has been suggested as the reason for the faster rolling velocities seen with L-selectin-mediated rolling compared with that mediated by E-selectin or P-selectin (33). The rolling velocity is set by kro, and thus the rolling velocity increase is due to the higher value for L-selectin. The state diagram shows, however, that γ must be smaller to maintain the rolling adhesive behavior on L-selectin.
Conclusions
In summary, our work illustrates how dynamic states of adhesion are controlled by bond physical chemistry (dissociative properties, association rates, and elasticity). Our method provides a way to predict the dynamics of adhesion from functional properties of the molecules. This work emphasizes the importance of two aspects of adhesion molecule physical chemistry: first, the need to measure kinetic rates, Bell model parameters, and elasticity accurately, and second, to understand how these functional properties are coded by structure. As more refined models are developed, they can be incorporated into the simulations. By addressing these problems, we will come to understand how structural motifs of different molecules are adapted for their functional roles in physiology and disease.
Acknowledgments
This work was supported by National Institutes of Health Grants HL18208 and GM59100.
Abbreviations
- PNAd
peripheral node addressin
- PSGL-1
P-selectin glycoprotein ligand 1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.200240897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.200240897
References
- 1.Springer T A. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Chen S, Springer T A. J Cell Biol. 1999;144:185–200. doi: 10.1083/jcb.144.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence M B, Springer T A. J Immunol. 1993;151:6338–6346. [PubMed] [Google Scholar]
- 4.Abbassi O, Kishimoto T K, McIntire L V, Anderson D C, Smith C W. J Clin Invest. 1993;92:2719–2730. doi: 10.1172/JCI116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg E L, McEvoy L M, Berlin C, Bargatze R F, Butcher E C. Nature (London) 1993;366:695–698. doi: 10.1038/366695a0. [DOI] [PubMed] [Google Scholar]
- 6.Brunk D K, Hammer D A. Biophys J. 1997;72:2820–2833. doi: 10.1016/S0006-3495(97)78924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alon R, Kassner P D, Carr M W, Finger E B, Hemler M E, Springer T A. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berlin C B, Bargatze R F, Campbell J J, von Andrian U H, Szabo M C, Hasslen S R, Nelson R D, Berg E L, Erlandsen S L, Butcher E C. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 9.Lawrence M B, Springer T A. Cell. 1991;65:859–874. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 10.Tempelman L A, Hammer D A. Biophys J. 1994;66:1231–1243. doi: 10.1016/S0006-3495(94)80907-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Andrian U H, Hasslen S R, Nelson R D, Erlandsen S L, Butcher E C. Cell. 1995;82:989–999. doi: 10.1016/0092-8674(95)90278-3. [DOI] [PubMed] [Google Scholar]
- 12.von Andrian U H, Chambers J D, McEvoy L M, Bargatze R F, Arfors K-E, Butcher E C. Proc Natl Acad Sci USA. 1991;88:7538–7542. doi: 10.1073/pnas.88.17.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S, Alon R, Fuhlbrigge R C, Springer T A. Proc Natl Acad Sci USA. 1997;94:3172–3177. doi: 10.1073/pnas.94.7.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunk D K, Goetz D J, Hammer D A. Biophys J. 1996;71:2902–2907. doi: 10.1016/S0006-3495(96)79487-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell G I. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 16.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dembo M, Torney D C, Saxman K, Hammer D. Proc R Soc London B. 1988;234:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]
- 18.Evans E. Faraday Disc. 1998;111:1–16. doi: 10.1039/a809884k. [DOI] [PubMed] [Google Scholar]
- 19.Evans E, Ritchie K. Biophys J. 1999;76:2439–2447. doi: 10.1016/S0006-3495(99)77399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammer D A, Apte S M. Biophys J. 1992;63:35–57. doi: 10.1016/S0006-3495(92)81577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alon R, Hammer D A, Springer T A. Nature (London) 1995;374:539–542. doi: 10.1038/374539a0. [DOI] [PubMed] [Google Scholar]
- 22.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Nature (London) 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 23.Fritz J, Katopodis A G, Kolbinger F, Anselmetti D. Proc Natl Acad Sci USA. 1998;95:12783–12288. doi: 10.1073/pnas.95.21.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang K-C, Hammer D A. Langmuir. 1996;12:2271–2282. [Google Scholar]
- 25.Kuo S C, Hammer D A, Lauffenburger D A. Biophys J. 1997;73:517–531. doi: 10.1016/S0006-3495(97)78090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang K-C, Hammer D A. Biophys J. 1999;76:1280–1292. doi: 10.1016/S0006-3495(99)77291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldman A J, Cox R G, Brenner H. Chem Eng Sci. 1967;22:653–660. [Google Scholar]
- 28.Lawrence M B, McIntire L V, Eskin S G. Blood. 1987;70:1284–1290. [PubMed] [Google Scholar]
- 29.Lawrence M B, Smith C W, Eskin S G, McIntire L V. Blood. 1990;75:227–237. [PubMed] [Google Scholar]
- 30.Morozov V N, Morozova T Y. Commun Mol Cell Biophys. 1990;6:249–270. [Google Scholar]
- 31.Goetz D J, El-Sabban M E, Pauli B U, Hammer D A. Biophys J. 1994;66:2202–2209. doi: 10.1016/S0006-3495(94)81016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.House S D, Lipowsky H H. Microvasc Res. 1991;42:288–304. doi: 10.1016/0026-2862(91)90063-h. [DOI] [PubMed] [Google Scholar]
- 33.Alon R, Chen S, Puri K D, Finger E B, Springer T A. J Cell Biol. 1997;138:1169–1180. doi: 10.1083/jcb.138.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith M J, Berg E L, Lawrence M B. Biophys J. 1999;77:3371–3383. doi: 10.1016/S0006-3495(99)77169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramachandran V, Nollert M U, Qiu H, Liu W-J, Cummings R D, Zhu C, McEver R P. Proc Natl Acad Sci USA. 1999;96:13771–13776. doi: 10.1073/pnas.96.24.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alon R, Chen S, Fuhlbrigge R, Puri K D, Springer T A. Proc Natl Acad Sci USA. 1998;95:11631–11636. doi: 10.1073/pnas.95.20.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strigl M, Simson D A, Kacher C M, Merkel R. Langmuir. 1999;15:7316–7324. [Google Scholar]
- 38.Heisig N. Adv Microcirc. 1968;1:89–94. [Google Scholar]
- 39.Atherton A, Born G V R. J Physiol. 1972;222:447–474. doi: 10.1113/jphysiol.1972.sp009808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alon R, Feizi T, Yuen C-T, Fuhlbrigge R C, Springer T A. J Immunol. 1995;154:5356–5366. [PubMed] [Google Scholar]
- 41.Varki A. J Clin Invest. 1997;99:158–162. doi: 10.1172/JCI119142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehta P, Cummings R D, McEver R P. J Biol Chem. 1998;273:32506–32513. doi: 10.1074/jbc.273.49.32506. [DOI] [PubMed] [Google Scholar]
- 43.Gittes F, Mickey B, Nettleton J, Howard J. J Cell Biol. 1993;120:923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson H P. Proc Natl Acad Sci USA. 1994;91:10114–10118. doi: 10.1073/pnas.91.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]