

Abstract
It has long been conjectured that the canonical genetic code evolved from a simpler primordial form that encoded fewer amino acids [e.g., Crick, F. H. C. (1968) J. Mol. Biol. 38, 367–379]. The most influential form of this idea, “code coevolution” [Wong, J. T.-F. (1975) Proc. Natl. Acad. Sci. USA 72, 1909–1912], proposes that the genetic code coevolved with the invention of biosynthetic pathways for new amino acids. It further proposes that a comparison of modern codon assignments with the conserved metabolic pathways of amino acid biosynthesis can inform us about this history of code expansion. Here we re-examine the biochemical basis of this theory to test the validity of its statistical support. We show that the theory's definition of “precursor-product” amino acid pairs is unjustified biochemically because it requires the energetically unfavorable reversal of steps in extant metabolic pathways to achieve desired relationships. In addition, the theory neglects important biochemical constraints when calculating the probability that chance could assign precursor-product amino acids to contiguous codons. A conservative correction for these errors reveals a surprisingly high 23% probability that apparent patterns within the code are caused purely by chance. Finally, even this figure rests on post hoc assumptions about primordial codon assignments, without which the probability rises to 62% that chance alone could explain the precursor-product pairings found within the code. Thus we conclude that coevolution theory cannot adequately explain the structure of the genetic code.
The apparent nonrandom distribution of amino acid assignments found within the canonical genetic code begs the question of what, if anything, code structure tells us about code evolution (e.g., refs. 1–4). One set of theories holds that natural selection has organized codon assignments to minimize the deleterious phenotypic impact of single nucleotide mutations and translational errors (4–7). When one calculates the average effect of single nucleotide substitutions for all codons under reasonable assumptions about mutation biases, the arrangement of amino acid assignments in the canonical genetic code conserves important amino acid properties better than almost all computer-generated alternatives (8–11).
A very different theory, called code coevolution, holds that the organization of the canonical code reflects the historical pathways by which amino acid biosynthesis evolved at the dawn of life (3, 12–20). The theory postulates that the earliest genetic code used a small number of prebiotically synthesized amino acids (21), and subsequently expanded to its present form by incorporating novel derivatives of these primordial amino acids as metabolic (biosynthetic) pathways evolved. Although the general idea of code expansion originated much earlier (1) and has appeared in many forms (see refs. 4, 20, 22, and 23 for selective reviews), coevolution theory has gained the widest acceptance by proposing an explicit pathway of code evolution that apparently had strong statistical support (3, 19, 20). Specifically, a central tenet of coevolution theory is that a “product” amino acid synthesized from a precursor amino acid usurped codons previously assigned to this precursor, such that the sequence of steps by which the code expanded is visible within modern codon assignments (Fig. 1).
Figure 1.

Evolutionary map of the genetic code based on precursor-product pairs in coevolution theory (adapted from ref. 3). Dashed boxes indicate putative primordial codon assignments required to create the relationships predicted by coevolution theory. Italicized codons do not match coevolution predictions even with this secondary assumption.
More than 100 publications have either built on or espoused the original coevolution model. These papers address topics including: the mechanism by which codons were ceded from precursor to product amino acids (see refs. 24–26 for overviews), the statistical basis for perceived coevolutionary patterns within the canonical code, the intermediate codes leading to the structure we see today (e.g., refs. 27 and 29), and evolutionary explanations for the expansion process (e.g., refs. 28 and 29). Indeed the claim that recent literature “has taken for granted the existence of a correlation between the biosynthetic relationships of amino acids and genetic code organization” (20) is legitimate; the only direct reappraisal of coevolution theory (30) demonstrated diminished statistical significance when different methods were used, but has itself been criticized on methodological grounds (refs. 19 and 20, but see ref. 31). Here we avoid methodological debate by using the analytical methods of the original coevolution paper (3). Instead we focus on the fundamental biochemical assumptions of the model. After identifying and correcting flaws in coevolution theory, we re-evaluate the statistical support for this biosynthetic theory of genetic code evolution.
Methodology
Coevolution theory defines a precursor amino acid as one in which any portion of the amino acid (backbone or side-chain) is metabolically incorporated into the product amino acid. The product is defined as the amino acid that lies the fewest number of metabolic steps from the precursor amino acid. However, the model excludes precursor-product pairs based on α-transaminations, because this reaction is particularly nonspecific and hence evolutionarily uninformative (3). The 13 precursor-product pairs described by these criteria are shown in Table 1.
Table 1.
Precursor-product pairs
| Glu→Arg | Asp→Asn | Ser→Trp | Thr→Ile | Val→Leu |
| Glu→Gln | Asp→Thr | Ser→Cys | Thr→Met | |
| Glu→Pro | Asp→Lys | Phe→Tyr | Gln→His | |
| Asp→Arg | Asp→Asn | Ser→Trp | Thr→Ile | |
| Glu→Gln | Asp→Thr | Ser→Cys | Asp→Met | |
| Glu→Pro | Asp→Lys | Phe→Tyr | Gln→His |
The 13 precursor-product pairs defined by coevolution theory (3) are shown in the first three lines. The revised list of 12 pairs based on a biochemically plausible definition of precursor-product pair is shown in the last three lines. Differences from coevolution theory are in bold.
Classically, statistical support for coevolution comes from applying the hypergeometric distribution to each precursor-product pair to calculate the probability that chance alone would cause the observed number of codons assigned to product amino acids to lie a single point mutation away from precursor codons. This probability is given by the equation:
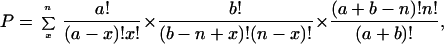 |
1 |
where a denotes the total number of codons lying one point mutation from the codons of the precursor amino acid, b denotes the number of codons that lie more than one point mutation from the codons of the precursor amino acid, x denotes the number of product codons that lie a single point mutation from the precursor codons (i.e., that fit the coevolution prediction), and n denotes the total number of product codons. In other words, this equation evaluates the combinatorial question: given a precursor amino acid's assignments within the code, what is the probability that random assignment of n product amino acid codons would produce x or more that fit the predictions of coevolutionary theory (i.e., lie within one point mutation of at least one precursor codon)?
Individual probabilities for each pair (Table 2) are then combined by Fisher's method to provide the overall probability that random codon assignments would produce patterns as consistent with coevolution theory as those of the canonical code (i.e., the sum of the −2 ln P values for the precursor-product pairs is distributed as χ2 with 2d degrees of freedom, where d denotes the number of pairs).§ When applied to the unambiguous precursor-product pairs of Table 1 this method produces χ2 = 44.76, df = 16 (Table 2), indicating a (one-tailed) probability of P = 0.00015¶ that the organization of the canonical code could result from chance (3). Thus, this selection of precursor-product pairs and methodology apparently provides strong statistical support for coevolutionary theory.
Table 2.
Statistical support for coevolution theory, adapted from ref. 3
| x | n | a | b | P(X ≥ x) | −2lnP | |
|---|---|---|---|---|---|---|
| Ser→Trp | 1 | 1 | 31 (34) | 24 (24) | 0.564 (0.59) | 1.15 (1.07) |
| Ser→Cys | 2 | 2 | 31 (34) | 24 (24) | 0.313 (0.34) | 2.32 (2.16) |
| Val→Leu | 6 | 6 | 24 (24) | 33 (36) | 0.00371 (0.00269) | 11.2 (11.84) |
| Thr→Ile | 3 | 3 | 24 (24) | 33 (36) | 0.069 (0.0591) | 5.34 (5.66) |
| Gln→His | 2 | 2 | 12 (14) | 47 (48) | 0.039 (0.0481) | 6.51 (6.07) |
| Phe→Tyr | 2 | 2 | 14 (14) | 45 (48) | 0.053 (0.0481) | 5.87 (6.07) |
| Glu→Gln | 2 | 2 | 12 (14) | 47 (48) | 0.039 (0.0481) | 6.51 (6.07) |
| Asp→Asn | 2 | 2 | 14 (14) | 45 (48) | 0.053 (0.0481) | 5.87 (6.07) |
All symbols are as per Eq. 1, with the addition of P(X ≥ x) used to denote the probability that chance alone would lead to x out of n product codons lying a single point mutation from the precursor amino acid. Parenthetic values include TER codons when calculating a and b. χ2 = 44.76 (45.00).
Criticism of Coevolution Theory
Using an alternative approach, Amirnovin (30) generated a large sample of randomized codes that maintained the synonymous block structure of the canonical code (Fig. 2c). By calculating the proportion of this sample that exhibited a higher codon correlation score (an additive measure of the number of product codons lying a single point mutation from corresponding precursor codons) for the set of precursor-product pairs in Table 1, he estimated a 0.1% probability that the patterns within the canonical code could result from chance. He further found that when the Gln→His pair and Val→Leu pair were eliminated from the set, this probability increased to 3.6%, and finally by drawing on the full web of amino acid biosynthetic relatedness from the metabolic pathways of Escherichia coli, he found that the probability rose to 34% that a randomly generated code would show a stronger pattern of biosynthetic relatedness than the canonical code. Although certain aspects of Amirnovin's method have been criticized (refs. 19 and 20 but see ref. 31), these same critical analyses (19, 20) confirmed that the choice of precursor-product pairs strongly influences the statistical significance of this result.
Figure 2.

Defining possible alternative codes: (a) the canonical genetic code; (b) coevolution theory assumes that any codon can take any amino acid assignment; (c) Amirnovin's critical reappraisal (30) assumes that redundancy patterns are fixed, and that only the identities of synonymous coding blocks can vary; and (d) biochemical considerations indicate that the translation machinery cannot distinguish codons that differ only by a U or C in the third position.
Defining Precursor-Product Amino Acid Pairs
Because the statistical significance of coevolution theory is so sensitive to the precise choice of precursor-product amino acid pairs, one can only make meaningful predictions if these pairs are based on unambiguous, consistent, and biologically relevant criteria. In fact, this is not the case for the pairs shown in Table 1.
Coevolution theory claims that the conserved pathways of amino acid biosynthesis in modern organisms (i.e., those found in all three domains of life) can be used to infer the historical precursor-product relationships between amino acids. The simplest criticism of coevolution theory is that some of the precursor-product pairs originally suggested do not conform to the theory's definition, which is based purely on the metabolic proximity of amino acids in terms of the number of enzyme-catalyzed steps in metabolic pathways (Fig. 3 a and b). For example, the Glu→Arg precursor-product pair is used (six enzymatic steps), despite the fact that proline and aspartate are, respectively, only five and two enzymatic steps away from arginine. In addition the precursor of methionine should be either cysteine (three enzymatic steps) or serine (three enzymatic steps, as it donates the methyl group to homocysteine through N5-methyl-5MeH4folate) rather than threonine (six steps) (32).
Figure 3.

Biosynthesis of (a) methionine: Asp is a valid precursor; Thr is not because it requires an energetically unfavorable conversion; (b) arginine: Asp lies two enzymatic steps away from Arg, whereas Pro is five and Glu is six enzymatic steps removed; and (c) leucine: Val is not a valid precursor of Leu because both are products of pyruvate in different enzymatic pathways.
Moreover the definition of precursor-product pairs is itself flawed because it incorporates several precursors, which do not actually precede their products in a metabolic pathway, but instead represent an alternative branch from a common intermediate (e.g., Thr→Met, Val→Leu) (32). This is a problem because the definition allows amino acids that lie on nonenergetically favorable pathways to be considered precursors of other amino acids.
In the case of the Thr→Met pair, coevolution infers that methionine would be synthesized from threonine through a homoserine intermediate (Fig. 3a). In the biosynthetic pathways of modern organisms this is not the case. Rather, methionine is synthesized from aspartate because the free energy change for synthesizing homoserine from threonine is prohibitive. Specifically the production of homoserine from threonine requires the reversal of two steps that are strongly favored because they are coupled to hydrolysis of ATP. Reversing these steps would require production of an ATP molecule from a lower energy ADP and Pi without a significant input of free energy. Under typical cellular conditions, the ΔG of ATP formation from ADP and Pi is 12 kcal/mol (33). Although the ΔG of formation for homoserine has not been reported, homoserine should exhibit a similar Gibbs free energy of formation to that of threonine because they are isomers (for comparison, the difference in free energy of formation between the isomers leucine and isoleucine is 0.1 kcal/mol) (34). Making the extremely conservative assumption that homoserine is 2 kcal/mol more stable than threonine, the free energy required to produce homoserine from threonine using existing metabolic pathways is 10 kcal/mol. This proposed biochemical pathway thus would give an equilibrium concentration of threonine that is 10 million times higher than homoserine. Because the amount of the homoserine intermediate synthesized from threonine would be insignificant to that synthesized from aspartate, it is difficult to see how threonine rather than aspartate could be considered the precursor for methionine. It is possible that primordial biosynthetic pathways not found in modern organisms could efficiently produce homoserine from threonine, but this argument lacks any supportive evidence and runs counter to coevolution's central claim that precursor-product pairs are reflected in the biosynthetic relationships of modern organisms.
Revised List of Precursor Product-Pairs
By excluding pairs for which the precursor does not share the same metabolic branch as the product, we define a self-consistent and biologically relevant set of precursors as the closest direct antecedents to their product amino acid in an energetically favorable non-α-transamination pathway. Under this definition, Val→Leu should be eliminated from the list of precursor-product pairs because they are produced from alternative branches of a common intermediate. In addition, the Thr→Met pair should be replaced by Ser→Met, Cys→Met, or Asp→Met, because the precursor molecules are all closer direct antecedents of methionine in energetically favorable pathways. However, the original coevolution paper claims that cysteine and serine should not be considered precursors of methionine, because other molecules could potentially contribute methionine's sulfur and methyl groups (3). Thus we have chosen to use the Asp→Met precursor-product pair. The revised list of precursor-product pairs is shown in Table 1. The use of this set of pairs reduces the statistical significance of patterns within the canonical code to P = 0.0062 (χ2 = 33.57, df = 16).
Notably, these revisions remain generous to coevolution theory. For example, we retain the Phe→Tyr pair despite the fact that prokaryotic biosynthesis of each amino acid proceeds along separate pathways from a common chorismate precursor. We have not eliminated this pair because Tyr is synthesized from Phe in a single step in its degradation pathway (32). In addition, we retain Gln as the precursor of His although it donates only a single nitrogen atom to this product. Removal of either of these relationships from calculations would further increase all quoted P values.
Biochemical Constraints on Alternative Codes
The extreme statistical significance originally reported for coevolution theory assumes that any amino acid may be assigned to any codon independently of all other codon assignments. In fact, there is a compelling biochemical reason to reject this assumption. No known tRNA anticodon base modification, natural or engineered, can discriminate third-base pyrimidines within any codon, despite the large number of nonstandard codes now recognized (35) and diverse experimental manipulation of coding components (e.g., see ref. 36). Quite simply, the translation machinery appears to read NNY codon blocks as synonymous by necessity. Ignoring this restriction significantly overestimates the probability of finding patterns of biosynthetic relatedness as strong as within the canonical code because of the combinatorial nature of the test statistics. Hence we examine this constraint.
By combining codons which differ by a U and C in their third position into synonymous blocks (Fig. 2d), the number of unique codon assignments is reduced from 61 to 45 and the number of alternative codes drops from 5.6 × 1064 to 2.8 × 1045. Although Wong considered this restriction in the original coevolution paper (3), it was claimed to have only a minimal impact on calculations of statistical significance. In fact, the exclusion of these biologically irrelevant genetic codes has a dramatic effect on the statistical significance of coevolution theory. The sum of the −2lnP values for the revised set of precursor-product pairs (Table 3), produces P = 0.168 (χ2 = 21.27, df = 16), a 16.8% probability that precursor-product pairs within the code could be explained as a chance artifact.
Table 3.
Probability that n product codon blocks lie a single point mutation away from the precursor codon blocks, where codons that differ by a U and C in the third position are constrained to code for the same amino acid
| x | n | a | b | P(X ≥ x) | −2lnP | |
|---|---|---|---|---|---|---|
| Ser→Trp | 1 | 1 | 21 (24) | 20 (20) | 0.512 (0.545) | 1.34 (1.21) |
| Ser→Cys | 1 | 1 | 21 (24) | 20 (20) | 0.512 (0.545) | 1.34 (1.21) |
| Phe→Tyr | 1 | 1 | 8 (8) | 36 (39) | 0.182 (0.170) | 3.41 (3.54) |
| Thr→Ile | 2 | 2 | 18 (20) | 24 (25) | 0.178 (0.192) | 3.46 (3.30) |
| Gln→His | 1 | 1 | 11 (11) | 32 (35) | 0.256 (0.239) | 2.73 (2.86) |
| Glu→Gln | 2 | 2 | 11 (13) | 32 (33) | 0.0609 (0.0754) | 5.60 (5.17) |
| Asp→Asn | 1 | 1 | 8 (8) | 36 (39) | 0.182 (0.170) | 3.41 (3.54) |
| Asp→Met | 0 | 1 | 8 (8) | 36 (39) | 1.00 (1.00) | 0.00 (0.00) |
χ2 = 21.27 (20.84).
Incorporating the Remaining Precursor-Product Pairs into the Probability Calculations
The 16.8% probability above is calculated from only eight of the 12 biochemically valid pairs. The original analysis excluded the remaining four pairs because they were based on two post hoc assumptions: (i) codons for glutamine were ceded from glutamate after glutamate had ceded codons to proline and arginine and (ii) codons for asparagine were ceded from aspartate after aspartate had ceded codons to lysine and threonine (3). Because there is no direct support for this scenario, no attempt was made to quantify the impact of these pairs on probability. However, it was claimed that their incorporation would decrease the P value (3).
Contrary to this claim, the incorporation of the precursor-products Glu→Arg, Glu→Pro, Asp→Thr, Asp→Lys into the analysis actually increases the P value. This is because many codons of the product amino acids lie more than a single point mutation from the precursor's codons, even accepting Wong's (3) putative primordial assignments. For example, only two of arginine's six codons lie a single point mutation from the putative primordial glutamate codons. Indeed a systematic incorporation of these pairs into the calculations (Table 4) leads to a probability of P = 0.232 (χ2 = 28.70, df = 24). Thus incorporation of all precursor-product pairs that meet a biochemically relevant definition and the restriction to genetic codes with NNY synonymous codon blocks shows that coevolution has little power to explain the structure of the genetic code.
Table 4.
Additional probabilities, as per Table 3, for the four legitimate precursor/product pairs omitted from the original analysis, accepting Wong's postulate (3) that codon blocks CAR were assigned to Glu and AAY assigned to Asp (see Fig. 1)
| x | n | a | b | P(X ≥ x) | −2lnP | |
|---|---|---|---|---|---|---|
| Asp→Lys | 2 | 2 | 12 (12) | 31 (34) | 0.0731 (0.0638) | 5.23 (5.50) |
| Glu→Pro | 2 | 3 | 16 (18) | 25 (26) | 0.334 (0.362) | 2.19 (2.03) |
| Asp→Arg | 0 | 5 | 12 (12) | 31 (34) | 1 (1) | 0 (0) |
| Asp→Thr | 1 | 3 | 12 (12) | 31 (34) | 0.636 (0.606) | 0.91 (1.00) |
If one rejects this postulate, then none of the product codons lies within one point mutation of the associated precursor codons [P(X ≥ x) = 1, −2lnP = 0 for all pairs]. Total χ2 = 28.70 + 8.42 (28.39 + 8.53).
Furthermore, if we reject these speculative assumptions about the identity and ordering of primordial codon assignments, and consider only the assignments of the standard genetic code (i.e., exclude the codons shown in dashed boxes in Fig. 1), the P value increases substantially: the nonoverlap of codons for Gln→Arg, Gln→Pro, Asn→Thr, Asn→Lys leads to an overall probability of P = 0.62 (χ2 = 21.27, df = 24) that the arrangement of precursor-product pairs observed within the code is a chance artifact.
Conclusions and Discussion
The statistical significance originally attributed to coevolution theory is caused by a number of inappropriate assumptions. First, several precursor-product amino acid pairs were based on implausible metabolic pathways, and pairs that fit the precursor-product definition (but not the predictions) of coevolution theory were ignored. Second, the model inappropriately defined the set of possible codes that could exist in the absence of code coevolution. Indeed, most of the statistical significance ascribed to coevolution theory stems from the invalid assumption that no other process could group individual amino acids into NNY codon blocks. Correction for these unambiguous errors leads to a 23% probability that a randomly generated code would display biosynthetic patterns as strong as or stronger than those found in the canonical code. Furthermore, this probability would increase to 62% if coevolution theory did not incorporate secondary assumptions about the historical order in which codons were reassigned from precursor to product amino acid. Thus the degree to which coevolution theory can explain the pattern of codon assignments in the canonical code is indistinguishable from a chance artifact: coevolution theory does not provide a satisfactory explanation of code structure.
It is important, however, to distinguish this finding from a general dismissal of code evolution through code expansion. Several independent lines of evidence suggest that the repertoire of coded amino acids grew during early evolution. For example, regardless of energy source and precise initial mixture of gases, prebiotic simulation experiments consistently predict that only a subset of the 20 proteinaceous amino acids were available at the dawn of life. This finding is strengthened by the remarkable overlap with studies of the composition of extraterrestrial debris such as the Murchison meteorite (20). Furthermore, many bacterial species code for certain prebiotically implausible amino acids in a manner that may reflect the process of code expansion, by enzymatically modifying a precursor amino acid attached to its cognate tRNA (see refs. 37–39 for recent overviews). Whether this is remnant of the expansion of a biochemical pathway remains unclear (but see ref. 38).
Several studies conclude that tRNA phylogeny independently supports coevolution theory (e.g., refs. 43–45). In fact, they present variable evidence that evolutionarily related tRNA species are associated with biosynthetically related amino acids in extant organisms: none provides the explicit picture of tRNA evolution needed to test coevolution's specific model (e.g., see ref. 45). Indeed, mutations that convert tRNAs between isoaccepting groups could obscure this history in modern genomes (46, 47).
It is also noteworthy that other patterns of biosynthetic relatedness have been reported within the code (e.g., refs. 40 and 41). Specifically, amino acids from the same biosynthetic pathway tend to be assigned to codons that begin with the same first base. Although the generality of this observation (lacking specific precursor/product pairs) removes problems with unrealistic pathways and lends strong statistical support (20), the pattern cannot be explained in terms of any known biological process. Furthermore, the lack of specific predictions about individual codon assignments appears to require the additional intervention of natural selection (for error minimization) to adequately explain the structure of the genetic code (10, 42).
With such considerations in mind, we word our conclusion with extreme care. Biochemical considerations and statistical reanalysis show that the product-precursor pairings at the heart of code coevolution theory are unreliable markers for a putative evolutionary process of code expansion. Subsequent analyses that used these pairings to predict the intermediate steps of code evolution, and to infer the evolutionary forces at work (27), are therefore speculative at best. It is plausible (if not probable) that the genetic code arose from a simpler form encoding fewer amino acids. However, it remains an open question whether any patterns of biosynthetic relatedness in the modern code can inform us of this process.
Acknowledgments
We thank Robin Knight, John Barratt, and Guy Sella for helpful discussion of this manuscript. S.J.F. was supported in part by grants from the Human Frontiers Science Program and the John Templeton Foundation.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250403097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250403097
Fisher's method assumes independence of observations. In fact, the calculations for each precursor-product pair are interdependent because fixing the assignments of each amino acid pair reduces the possible assignments for those that remain. The exact magnitude of this error cannot be calculated in the absence of a specific proposed order in which amino acids were assigned to codons: no proponent of coevolution has made such a proposal. Randomizing for all possible orderings, the average effect of this inaccuracy is that low quoted P values represent underestimates.
All quoted results exclude the three termination codons from calculations. Although the original analysis included termination codons (3), this has later been considered incorrect (20). In fact, the inclusion or exclusion of termination codons makes very little difference to individual probabilities. All tables show values based on exclusion followed by parenthetic values based on inclusion.
References
- 1.Crick F H C. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 2.Dillon L S. Bot Rev. 1973;39:301–345. [Google Scholar]
- 3.Wong J T-F. Proc Natl Acad Sci USA. 1975;72:1909–1912. doi: 10.1073/pnas.72.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knight R D, Freeland S J, Landweber L F. Trends Biochem Sci. 1999;24:241–247. doi: 10.1016/s0968-0004(99)01392-4. [DOI] [PubMed] [Google Scholar]
- 5.Sonneborn T M. In: Evolving Genes and Proteins. Bryson V, Vogel H J, editors. New York: Academic; 1965. pp. 97–166. [DOI] [PubMed] [Google Scholar]
- 6.Zuckerkandl E, Pauling L. In: Evolving Genes and Proteins. Bryson V, Vogel H J, editors. New York: Academic; 1965. pp. 377–400. [DOI] [PubMed] [Google Scholar]
- 7.Woese C R. Proc Natl Acad Sci USA. 1965;54:1546–1552. doi: 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haig D, Hurst L D. J Mol Evol. 1991;33:412–417. doi: 10.1007/BF02103132. [DOI] [PubMed] [Google Scholar]
- 9.Freeland S J, Hurst L D. J Mol Evol. 1998;47:238–248. doi: 10.1007/pl00006381. [DOI] [PubMed] [Google Scholar]
- 10.Freeland S J, Knight R D, Landweber L F, Hurst L D. Mol Biol Evol. 2000;17:511–518. doi: 10.1093/oxfordjournals.molbev.a026331. [DOI] [PubMed] [Google Scholar]
- 11.Ardell D H. J Mol Evol. 1998;47:1–13. doi: 10.1007/pl00006356. [DOI] [PubMed] [Google Scholar]
- 12.Wong J T-F. Proc Natl Acad Sci USA. 1976;73:2336–2340. doi: 10.1073/pnas.73.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong J T-F. Trends Biochem Sci. 1981;6:33–35. [Google Scholar]
- 14.Wong J T-F. Microbiol Sci. 1988;5:174–181. [PubMed] [Google Scholar]
- 15.DiGiulio M. J Mol Evol. 1989;29:191–201. [Google Scholar]
- 16.DiGiulio M. Origins Life Evol Biosphere. 1994;25:549–564. [Google Scholar]
- 17.DiGiulio M. Origins Life Evol Biosphere. 1996;26:589–609. doi: 10.1007/BF01808222. [DOI] [PubMed] [Google Scholar]
- 18.DiGiulio M. J Mol Evol. 1998;46:615–621. [Google Scholar]
- 19.DiGiulio M. J Mol Evol. 1999;48:253–255. doi: 10.1007/pl00006464. [DOI] [PubMed] [Google Scholar]
- 20.DiGiulio M, Medugno M. J Mol Evol. 2000;50:258–263. doi: 10.1007/s002399910030. [DOI] [PubMed] [Google Scholar]
- 21.Wong J T-F, Bronskill P M. J Mol Evol. 1979;13:115–125. doi: 10.1007/BF01732867. [DOI] [PubMed] [Google Scholar]
- 22.DiGiulio M. J Theor Biol. 1997;187:573–581. doi: 10.1006/jtbi.1996.0390. [DOI] [PubMed] [Google Scholar]
- 23.Judson O P, Haydon D. J Mol Evol. 1999;49:539–550. doi: 10.1007/pl00006575. [DOI] [PubMed] [Google Scholar]
- 24.DiGiulio M. J Theor Biol. 1998;191:191–196. [Google Scholar]
- 25.Szathmary E. Trends Genet. 1999;15:223–229. doi: 10.1016/s0168-9525(99)01730-8. [DOI] [PubMed] [Google Scholar]
- 26.Davis B K. Prog Biophys Mol Biol. 1999;72:157–243. doi: 10.1016/s0079-6107(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 27.DiGiulio M, Medugno M. J Mol Evol. 1999;49:1–10. doi: 10.1007/pl00006522. [DOI] [PubMed] [Google Scholar]
- 28.Edwards M R. J Theor Biol. 1996;179:313–322. doi: 10.1006/jtbi.1996.0070. [DOI] [PubMed] [Google Scholar]
- 29.Popa R. J Mol Evol. 1997;44:121–127. doi: 10.1007/pl00006128. [DOI] [PubMed] [Google Scholar]
- 30.Amirnovin R. J Mol Evol. 1997;44:473–476. doi: 10.1007/pl00006170. [DOI] [PubMed] [Google Scholar]
- 31.Amirnovin R, Miller S L. J Mol Evol. 1999;48:254–255. [Google Scholar]
- 32.Voet D, Voet J G, Pratt C W. Fundamentals of Biochemistry. New York: Wiley; 1999. [Google Scholar]
- 33.Stryer L. Biochemistry. New York: Freeman; 1995. [Google Scholar]
- 34.Hutchens J O. In: Handbook of Biochemistry and Molecular Biology, Physical, and Chemical Data. 3rd Ed. Fasman G D, editor. Cleveland: CRC; 1976. pp. 111–113. [Google Scholar]
- 35.Knight, R. D., Freeland, S. J. & Landweber, L. F. (2000) Nat. Rev. Genet., in press. [DOI] [PubMed]
- 36.Söll D, RajBhandary U L. tRNA: Structure, Biosynthesis, and Function. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 37.Becker H D, Kern D. Proc Natl Acad Sci USA. 1998;95:12832–12837. doi: 10.1073/pnas.95.22.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown J R, Doolittle W F. J Mol Evol. 1999;49:485–495. doi: 10.1007/pl00006571. [DOI] [PubMed] [Google Scholar]
- 39.Blanquet S, Mechulam Y, Schmitt E. Curr Opin Struct Biol. 2000;10:95–101. doi: 10.1016/s0959-440x(99)00055-x. [DOI] [PubMed] [Google Scholar]
- 40.Miseta A. Physiol Chem Phys Med NMR. 1989;21:237–242. [PubMed] [Google Scholar]
- 41.Taylor F J R, Coates D. BioSystems. 1989;22:177–187. doi: 10.1016/0303-2647(89)90059-2. [DOI] [PubMed] [Google Scholar]
- 42.Freeland S J, Hurst L D. Proc R Soc London Ser B. 1998;265:2111–2119. [Google Scholar]
- 43.Bermudez C I, Daza E E, Andrade E. J Theor Biol. 1999;197:193–205. doi: 10.1006/jtbi.1998.0866. [DOI] [PubMed] [Google Scholar]
- 44.Chaley M B, Korotkov E V, Phoenix D A. J Mol Evol. 1999;48:168–177. doi: 10.1007/pl00006455. [DOI] [PubMed] [Google Scholar]
- 45.DiGiulio M. Origins Life Evol Biosphere. 1995;25:549–564. doi: 10.1007/BF01582024. [DOI] [PubMed] [Google Scholar]
- 46.Saks M E, Sampson J R. J Mol Evol. 1995;40:509–518. doi: 10.1007/BF00166619. [DOI] [PubMed] [Google Scholar]
- 47.Saks M E, Sampson J R, Abelson J. Science. 1998;279:1665–1670. doi: 10.1126/science.279.5357.1665. [DOI] [PubMed] [Google Scholar]