

Abstract
Archaeological, anatomical, linguistic, and genetic data have suggested that there is an old and significant boundary between the populations of north and south China. We use three human genetic marker systems and one human-carried virus to examine the north/south distinction. We find no support for a major north/south division in these markers; rather, the marker patterns suggest simple isolation by distance.
Archaeological and genetic evidence has suggested that the human population experienced a dramatic expansion in the last 100,000 years, spreading rapidly to its current worldwide area of occupation. Much of the area inhabited by humans must have been reached via migration through East Asia. There is evidence of human occupation through Central Asia to Beringia during the later Pleistocene (1, 2) and parallel evidence of early humans in Australia and Southern Asia. One natural hypothesis is that the population of East Asia is a result of ancient contact and mixing between these northern and southern pincers of the modern human expansion. Alternatively, East Asia may have been reached by either a northern or southern route, followed by dispersal into nearby areas.
In separate analyses, patterns of dentition, archaeological assemblage composition, linguistics, familial surnames, and various low-resolution genetic systems have identified systematic differences between northern and southern groups (3–8) (see ref. 9 for a review). These lines of evidence have been taken as support for a strong north/south distinction that would appear to support the pincer model of the origin of East Asians. However, two recent high-resolution approaches to East Asian genetic diversity have come to different conclusions.
Chu et al. (10) and Su et al. (11) examined nuclear microsatellites and Y-chromosome nucleotide polymorphisms, respectively. Consistent with earlier work, patterns of diversity in microsatellite loci were found to fall into northern and southern clusters, with northern groups being polyphyletic. Y-chromosome polymorphisms found in northern populations were a perfect subset of those in the south—every Y-chromosome haplotype observed in northern groups was observed in at least one southern group, but not every lineage observed in southern groups was observed in a northern group. On the basis of these findings, Chu et al. and Su et al. argued that northern East Asian populations are derived from southern East Asian populations.
In this paper, we combine new evidence from mtDNA polymorphisms and five short tandem repeat (STR) loci and previously published evidence from Y-chromosome polymorphisms to reexamine the hypothesis that northern and southern China are distinct. We apply the same method to all loci, providing a simple basis for comparison, and contrast patterns of diversity in these markers with patterns in the distribution of JC virus, an asymptomatic urinary tract virus that is frequently transmitted from parent to child and that may provide information about human migrations (12, 13).
Materials and Methods
Data.
A dataset composed of four different marker types was assembled.
First, a total of 473 comparable mtDNA restriction fragment length polymorphism profiles was collected. High-resolution restriction endonuclease mapping of 113 individuals from four ethnic groups in southwest China (Bai, Dai, Lisu, and Yi) was performed according to the protocols of Torroni et al. (14), and analogous profiles from 360 individuals were collected from previously published papers to provide a basis for comparison (Fig. 1, Table 1). Data assembled from the literature included Ewenki (15), Korean (16), Malay Chinese (16), Nivikh (15), Taiwan Han (16), Tibetan (17), Udegey (15), Malay (16), Malay Aborigine (16), Sabah Aborigine (16), and Vietnamese (16) (Fig. 1).
Figure 1.

Map of sampled locations for mtDNA and STR loci. Data for other loci were assembled entirely from earlier studies (see Materials and Methods).
Second, five STR loci (12-nt repeats in the Exo I region of Dopamine Receptor 4; 48-nt repeats in the Exo III region of Dopamine Receptor 4; 120-nt repeats in the Pro region of Dopamine Receptor 4; 47-nt repeats in the 7q subtelomere region; 49-nt repeats in the 7q subtelomere region) were scored for repeat number in a total of 900 haploid genomes from 14 populations (Table 1).
Finally, 192 JC virus DNA sequences (13) from 17 populations and biallelic marker data from 836 Y chromosomes from 30 populations (11) were obtained from the literature (Table 1). Data on length polymorphisms in the Y chromosome were not included.
Populations were divided into northern and southern groups on the basis of a consensus of geography and documented history. For example, historical documents indicate that among the groups sampled in southwest China, the Dai is originally from southeastern areas, and the Yi, Bai, and Lisu are from areas farther north, so the populations were divided accordingly, although all of them were sampled in Yunnan Province (18).
Analysis.
We examined patterns of regional association by computing principal components of the population gene differences and plotting the first two principal components (19). The pictures are the best, in the sense of least-squares two-dimensional representation of genetic distances among populations, where the squared distance between population x and y is the sum over all sites of
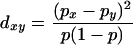 |
Here, px and py are the frequencies of a genetic variant in populations x and y, and p is the overall mean frequency of the variant.
Variation in mtDNA or the Y chromosome is often studied by computing an estimate of the whole genealogy of a sample of genes, a gene tree, and then interpreting the tree in terms of geography or population phylogeny. Details from these reconstructions may lead to appealing interpretations, but often little is known about statistical support for the interpretations. Gene trees are embedded in population histories, but it is not so clear how to read the population history from the gene tree, nor how to predict a gene tree from a postulated population history. Instead, we use principal components, in an exploratory spirit, as a simple visual summary of patterns of population difference.
Results and Discussion
Recent investigations by using molecular markers to study patterns of genetic diversity in East Asia have tried to address two main questions. First, are northern and southern East Asian populations genetically distinct? Second, are northern and southern East Asian populations descendants of the same ancestral population, or are they descended from different populations?
Chu et al. inferred a distinction between southern and northern Chinese populations and a southern origin for northerners by analyzing phylogenetic trees of populations constructed by using microsatellite data (10). Among trees constructed by using the neighbor-joining method with bootstrapping, Chu et al. identified a “clear distinction between southern and northern Chinese populations” on the basis of the presence of a paraphyletic northern group and an almost monophyletic southern clade. Su et al. inferred support for regional clusters on the basis of an analysis of principal components of Y-chromosomal diversity and the observation of substantial lineage sharing among regions, also suggesting the possibility of a southern origin (11). In a map of the first two principal components of variance, northern populations clustered together, and southern populations clustered together. In contrast, we find support for neither a strong regional distinction nor a southern origin of Northeast Asian populations.
Four sections in Fig. 2 show the results from our four marker systems. In these maps, the first principal component accounted for 11, 34, 33, and 32% of the total dispersal in observed in mtDNA, STR, Y chromosome, and JC virus, respectively. The second principal component accounted for 10, 19, 23, and 14% of the variance in observed in mtDNA, STR, Y chromosome, and JC virus, respectively. These maps suggest an alternative explanation for diversity patterns in East Asia. Three features of the principal components maps in Fig. 2 are most important.
Figure 2.

Principal components maps. For each map, the x axis is the first and the y axis the second principal component. Northern populations are indicated by open and southern populations by closed circles.
First, the STRs, the Y polymorphisms, and the JC virus polymorphisms are geographically structured so that the principal components give a good portrayal of the underlying genetic distances. There is, on the other hand, almost no structure in the mtDNA differences among regions. The genetic distances among populations from mtDNA describe something like a high-dimension sphere. Even though it is possible to generate an mtDNA phylogeny from the data, any such phylogeny could not reveal much of interest about population history here.
Second, although northern and southern populations generally fall into different regions of the principal components maps, the clusters are not distinct. For example, in the map of diversity in mtDNA, some southern populations such as Dai are much more similar to other northern populations than they are to other southerners, such as the Vietnamese or Malay Aborigines. The putative northern and southern clusters appear to blend across a cline; there is no abrupt change.
Third, populations sampled from adjacent geographical areas tend to be near each other on the principal components maps. This finding is consistent with previously published evidence of genetic isolation by distance in China (20) and explains the lack of clustering in the principal components maps. Because populations are isolated by distance, and because they were sampled on a predominantly north/south axis, the principal components maps can be segregated into northern and southern groups delineated by latitude. This feature explains the gradual, rather than sharp, divide among the arbitrarily chosen north/south divide.
The geographical organization of the principal components maps raises questions about the informativeness of human population “phylogenies.” If populations that are isolated by distance are sampled along an axis, is artificial phylogenetic signal introduced? One possible explanation for the repeated identification of northern and southern clades in East Asia (9, 10) is that oversampling along a north/south axis has spuriously influenced phylogenetic inferences. Such a bias would be consistent with the low bootstrap values supporting northern and southern clades observed by Chu et al. (10).
It is of some interest that the clearest north/south distinction among the four principal components maps in Fig. 2 is observed in the JC virus. The JC virus is a nonpathogenic urinary tract virus that is thought to be largely vertically transmitted, and as such it might be a replicate of mtDNA. However, the true extent of horizontal transmission in JC virus is unknown. Diversity patterns in the JC virus have been interpreted as evidence for a north/south distinction previously (21), and that the virus displays a stronger regional distinctiveness than the human genes suggests appreciable horizontal transmission may be present. The level of isolation by distance is more different among viral subpopulations than among their human hosts.
The lack of regional clusters brings inferences about directional migration into question as well. Su et al. suggest that the presence of every northern lineage in at least one southern population implies northward movement, but it is unreasonable to conclude that the northern population derived from the southern populations based on this evidence. One attractive alternative explanation for the asymmetry in lineage sharing is that northern and southern East Asian groups have had a long history of separation, but many lineages have migrated from north to south recently. Such asymmetric migration could easily generate regional differences in genetic diversity. However, this explanation would predict a clear genetic difference between northern and southern groups. Another explanation for the asymmetry in lineage sharing is suggested by regional differences in demography. Whereas southern populations reside mostly in high-density areas, northern areas are sparsely populated (22). Between-region migration, accompanied by high rates of genetic drift and lineage loss in northern groups, could account for an asymmetry in lineage composition without causing appreciable between-region divergence.
The potentially important role of Central Asia in questions about the genetic composition of East Asia is emphasized by patterns of mtDNA diversity in the region. In a comparison of mtDNA sequences from Europe, Central Asia, and East Asia, Comas et al. (23) found a closer affiliation between Mongols and Talas Kirghiz populations than between the Talas Kirghiz and Sarytash Kirghiz. No southern East Asians were included in Comas et al.'s study, and their relationships to Central Asians are unknown. However, the similarity of some northern East Asian populations to Central Asians indicates that the large migrations associated with trade along the Silk Road and during later times may have had an influence on diversity in the Far East. The inclusion of Central Asian samples in future studies including both northern and southern East Asians will be important in answering more detailed questions about East Asian origins.
The existence of a genetic distinction between northern and southern East Asia is not well supported. Patterns of genetic diversity in the area are more consistent with the notion that local gene flow since the end of the Pleistocene era has erased old human population differences over much of the world at neutral marker loci (24), and that much of the differentiation in the region is attributable to simple isolation by distance (20). Such erasure is expected to happen even if migration rates are relatively low (25). The lack of pattern in East Asia suggests that many of the anthropological trends previously held to define pervasive regional distinction are strictly cultural phenomena with no implications for genetic differentiation. This finding is itself interesting—regional cultural trends in East Asia seem to have persisted for lengthy time periods despite evident genetic continuity.
Table 1.
Sampled populations

Acknowledgments
We are indebted to Dr. Antonio Torroni for his detailed explanation of both experimental procedures and statistical methods. We thank Dr. Wen Wang and Long Nie for their intellectual contribution and support in sample collection. We also thank Xue-Mei Lu and Jing Luo for their comments on an early version of this paper. This work was supported by the Chinese Academy of Sciences, Natural Science Foundation of Yunnan Province, and National Sciences Foundation of China.
Abbreviation
- STR
short tandem repeat
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.240441297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.240441297
References
- 1.Goebel T. Evol Anthropol. 1999;8:208–227. [Google Scholar]
- 2.Davis R S, Ranov V A. Evol Anthropol. 1999;8:186–193. [Google Scholar]
- 3.Turner C G I. Am. J. Phys. Anthropol. 1987. 305–321. [DOI] [PubMed] [Google Scholar]
- 4.Barnes G L. The Rise of Civilization in East Asia: The Archaeology of China, Korea and Japan. London: Thames and Hudson; 1999. [Google Scholar]
- 5.Ruhlen M. The Origin of Language. New York: Wiley; 1994. [Google Scholar]
- 6.Du R, Yuan Y, Huang J, Mountain J, Cavalli-Sforza L L. Journal of Chinese Linguistics Monograph Series. Oxford, U.K.: Oxford Univ. Press; 1992. [Google Scholar]
- 7.Matsumoto H. Hum Genet. 1988;80:207–218. doi: 10.1007/BF01790088. [DOI] [PubMed] [Google Scholar]
- 8.Zhao T M, Lee T D. Hum Genet. 1989;83:101–110. doi: 10.1007/BF00286699. [DOI] [PubMed] [Google Scholar]
- 9.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton Univ. Press; 1994. [Google Scholar]
- 10.Chu J Y, Huang W, Kuang S Q, Wang J M, Xu J J, Chu Z T, Yang Z Q, Lin K Q, Li P, Wu M, Geng Z C, et al. Proc Natl Acad Sci USA. 1998;95:11763–11768. doi: 10.1073/pnas.95.20.11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su B, Xiao J, Underhill P, Deka R, Zhang W, Akey J, Huang W, Shen D, Lu D, Luo J, et al. Am J Hum Genet. 1999;65:1718–1724. doi: 10.1086/302680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunitake T, Kitamura T, Guo J, Taguchi F, Kawabe K, Yogo Y. J Clin Microbiol. 1995;33:1448–1451. doi: 10.1128/jcm.33.6.1448-1451.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugimoto C, Kitamura T, Guo J, Al-Ahdal M N, Shchelkunov S N, Otova B, Ondrejka P, Chollet J Y, El-Safi S, Ettayebi M, et al. Proc Natl Acad Sci USA. 1997;94:9191–9196. doi: 10.1073/pnas.94.17.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torroni A, Schurr T G, Yang C C, Szathmary E J, Williams R C, Schanfield M S, Troup G A, Knowler W C, Lawrence D N, Weiss K M, Wallace D C. Genetics. 1992;130:153–162. doi: 10.1093/genetics/130.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torroni A, Sukernik R L, Schurr T G, Starikovskaya Y B, Cabell M F, Crawford M F, Comuzzie A G, Wallace D C. Am J Hum Genet. 1993;53:591–608. [PMC free article] [PubMed] [Google Scholar]
- 16.Ballinger S W, Schurr T G, Torroni A, Gan Y Y, Hodge J A, Hassan K, Chen K H, Wallace D C. Genetics. 1992;130:139–152. doi: 10.1093/genetics/130.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torroni A, Miller J A, Moore L G, Zamudio S, Zhuang J, Droma T, Wallace D C. Am J Phys Anthropol. 1994;93:189–199. doi: 10.1002/ajpa.1330930204. [DOI] [PubMed] [Google Scholar]
- 18.Du R, Vincent F Y. Ethnic Groups in China. Beijing: Science Press; 1993. [Google Scholar]
- 19.Harpending H C, Jenkins T. In: Method and Theory in Anthropological Genetics. Crawford M, Workman P, editors. Albuquerque, NM: Univ. New Mexico Press; 1973. pp. 177–199. [Google Scholar]
- 20.Chen K. Bull Ethnogr Acad Sin. 1992;73:209–232. [Google Scholar]
- 21.Guo J, Sugimoto C, Kitamura T, Ebihara H, Kato A, Guo Z, Liu J, Zheng S P, Wang Y L, Na Y Q, et al. J Gen Virol. 1998;79:2499–2505. doi: 10.1099/0022-1317-79-10-2499. [DOI] [PubMed] [Google Scholar]
- 22.m. Hsieh C, Hsieh J K. China: A Provincial Atlas. New York: Macmillan; 1995. [Google Scholar]
- 23.Comas D, Calafell F, Mateu E, Pérez-Lezaun A, Bosch E, Martínez-Arias R, Clarimon J, Facchini F, Fiori G, Luiselli D, et al. Am J Hum Genet. 1998;63:1824–1838. doi: 10.1086/302133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harpending, H. C. & Rogers, A. R. (2000) Annu. Rev. Genom. Hum. Genet.1, in press. [DOI] [PubMed]
- 25.Hartl D L, Clark A G. Principles of Population Genetics. 3rd Ed. Sunderland, MA: Sinauer; 1997. [Google Scholar]