

Abstract
Aims: Reperfusion of ischaemic myocardium after acute myocardial infarction (AMI) can induce ischaemia/reperfusion (I/R) injury, as a result of local activation of the complement system. C reactive protein (CRP) is involved in this activation. This study analysed the potential role of IgM in complement activation in the infarcted human myocardium.
Methods: Immunochemical analysis was carried out on heart specimens from 59 patients who died from AMI. Serial slides of frozen tissue from the infarction site were stained for IgM, complement factors C3d and C5b–9 (membrane attack complex), and CRP.
Results: IgM deposits were found on the plasma membrane, cross striations, and in the cytoplasm of jeopardised cardiomyocytes in infarcts of one to five days duration. IgM depositions were remarkably similar to those of CRP and both complement factors. The relative staining intensities of IgM and CRP varied greatly among patients.
Conclusions: Similar to CRP, IgM targets complement locally to jeopardised cardiomyocytes in the human heart after AMI. Localisation patterns and relative staining intensities suggest that IgM and CRP recognise similar epitopes in the ischaemic heart, but that the relative contribution of each protein to complement activation in the ischaemic myocardium differs among patients.
Keywords: IgM, immunology, inflammation, myocardial infarction
Reperfusion of the impaired myocardium after acute myocardial infarction (AMI) results in a local inflammatory response.1 This inflammatory response damages the ischaemic tissue, a phenomenon also designated ischaemia/reperfusion (I/R) injury. Prevention of this I/R induced inflammation has been shown to reduce the infarct size by as much as 50% in animal models,1 and may offer new therapeutic opportunities for patients with AMI. Hence, knowledge of the mechanisms of I/R injury in humans is warranted.
The complement system is an important mediator involved in experimental I/R injury in animals. In a rat model of reperfusion injury of ischaemic myocardium, prevention of complement activation resulted in a pronounced reduction of I/R related injury.2 In addition, there is preliminary evidence that the inhibition of complement by C1 inhibitor reduces infarction size by up to 57% in humans.3 These results point to complement as an attractive target for limiting I/R injury. However, the molecular basis of I/R induced complement activation is not completely understood.
“Prevention of ischaemia/reperfusion induced inflammation has been shown to reduce the infarct size by as much as 50% in animal models, and may offer new therapeutic opportunities for patients with acute myocardial infarction”
Various molecules have been claimed to target activated complement to the ischaemic myocardium during I/R injury. One of these molecules is C reactive protein (CRP), which activates complement via the classical pathway.4–6 In rabbits, CRP was localised to the inflamed myocardium after AMI.7,8 Administration of human CRP in rats challenged with coronary artery occlusion enhances infarction size in a complement dependent manner.9 In myocardial tissue specimens from patients that died from AMI, CRP colocalises with complement,10 suggesting that in humans this acute phase protein contributes to complement activation in the ischaemic myocardium. This notion is supported by observations that during AMI the human heart contains increased amounts of activation products that specifically reflect complement activation induced by CRP.11
In mice, IgM was also shown to be involved in complement activation induced during I/R injury.12,13 In I/R models of the intestine and skeletal muscle, IgM deficient mice developed substantially less I/R injury than their wild-type littermates, and this injury was restored in the deficient mice by supplementation with normal murine IgM. The specificity of the IgM mediating I/R injury in mice is not known. It is also not known whether a similar IgM dependent mechanism occurs in humans during I/R. We hypothesised that in humans IgM might also contribute to ischaemic injury in the heart after AMI. Therefore, in our study we analysed tissue specimens from the hearts of patients who had died from AMI. These specimens were analysed for the presence of IgM, CRP, complement factor C3d, and complement factor C5b–9 of the membrane attack complex.
METHODS
Patients
Patients referred to the department of pathology, VU Medical Centre, Amsterdam, the Netherlands for necropsy were included in our study if this was performed within 24 hours of death, and when at necropsy they showed signs of a recently developed AMI—that is, on histochemical examination they had decreased lactate dehydrogenase staining (decoloration) of the affected myocardium. Our study was approved by the ethics committee of the VU Medical Centre, Amsterdam, and complied with the principles of the Declaration of Helsinki. Use of leftover material after pathological examination is part of the standard patient contract in our hospital.
Processing of tissue specimens
Myocardial tissue specimens were obtained from the infarcted zone and from remote sites of the healthy part of the heart. These remote sites showed normal lactate dehydrogenase staining patterns and were studied as internal non-infarcted controls. A control heart tissue sample from the left ventricle was obtained from a patient who died from a cause not related to heart disease. Before being prepared as cryosections, tissue specimens were stored at −196°C (liquid N2). Frozen sections were mounted on to SuperFrost®Plus glass slides (Menzel-Gläser, Braunschweig, Germany).
Assessment of infarct phase
Microscopic criteria14,15 were used to estimate infarct duration and viability of cardiomyocytes in all myocardial tissue specimens. Because morphological judgment is more reliable with paraffin wax embedded slides, corresponding paraffin wax slides were also made. Jeopardised myocardium was characterised by the intensity of eosinophilic staining of involved myofibres, condensation, loss of nuclei, and cross striation. We characterised jeopardised myocardium without microscopic changes but with macroscopic lactate dehydrogenase decolorisation as an early phase infarct (phase 1), infiltration of polymorphonuclear leucocytes (PMNs) as a PMN phase infarct (phase 2), and infiltration of lymphocytes and macrophages and fibrosis as a chronic phase infarct (phase 3). Furthermore, patients showing typical changes of phase 3 morphology together with those of phase 1 morphology were classified as reinfarct early phase (phase 4). Patients with phase 3 morphology and phase 2 morphology were classified as reinfarct PMN phase (phase 5). Two investigators (PAJK and HWMN) judged and scored independently all slides for the infarct phase. Table 1 shows the distribution of the various infarct phases among the patients. In all cases, infarct age as assessed by histology corresponded with the clinical course.
Table 1.
Distribution of various phases of AMI in the patients
| AMI phase | Infarct age | Number of patients | Male/female | Age range |
| Early phase | 0–12 hours | 27 | 17/10 | 23–98 |
| PMN phase | 12 hours to 5 days | 11 | 6/5 | 42–85 |
| Chronic phase | 5–14 days | 5 | 3/2 | 58–83 |
| Reinfarction | Chronic phase + | |||
| Early phase | 0–12 hours | 5 | 3/2 | 63–85 |
| PMN phase | 12 hours to 5 days | 7 | 3/4 | 30–89 |
AMI, acute myocardial infarction; PMN, polymorphonuclear leucocyte.
Antibodies
Horseradish peroxidase conjugated rabbit polyclonal antihuman IgM antibody (American Qualex, San Clemente, California, USA) was used for immunohistochemical detection of IgM. Monoclonal antibodies (mAbs) against the complement factor C3d (mAb C3-15; subtype, IgG1) and against CRP (mAb 5G4; subtype, IgG2a) have been used previously for immunohistochemical studies.10 mAb aE11 (subtype, IgG2a; Dako, Carpinteria, California, USA) was used for the detection of complement factor C5b–9. The mAbs were stored at 1 mg/ml in phosphate buffered saline (PBS), pH 7.4. Irrelevant mAbs (two IgG1, one IgG2a, and one IgM) were used as negative controls, and tested at concentrations similar to those used for the anti-complement and anti-CRP mAbs. These controls yielded negative results.
Immunohistochemistry
Frozen sections (5 μm thick) were mounted on to glass slides, dried for one hour by exposure to air, and fixed in acetone (“Baker analysed reagent”; Mallinckrodt Baker, Deventer, the Netherlands). After rinsing in PBS, the slides were incubated at room temperature for 10 minutes with normal swine serum (for IgM), normal rabbit serum (for complement and CRP) (both Dakopatts, Glostrup, Denmark), or 5% (wt/vol) bovine serum albumin (BSA; Boehringer, Mannheim, Germany) in PBS (for C5b–9), diluted 1/10 (normal swine serum) or 1/50 (normal rabbit serum) in 1% (wt/vol) BSA in PBS (PBS-BSA). Incubation of the slides with specific antibody solutions (diluted in PBS-BSA) was performed for 60 minutes except for mAb aE11, which was incubated overnight at 4°C (the polyclonal antibody against IgM was diluted 1/400; mAb C3-15 was diluted 1/1500; mAb 5G4 was diluted 1/300; mAb aE11 was diluted 1/50). The slides were washed for 30 minutes with PBS and slides stained with mAbs were incubated with horseradish peroxidase conjugated rabbit antimouse immunoglobulins (Dakopatts), diluted 1/25 in PBS-BSA except for mAb aE11, which was detected using EnVision (Dako). Thereafter, the slides were washed again in PBS and incubated for three minutes in 0.5 mg/ml 3,3′-diaminobenzidine tetrahydrochloride (Sigma, St Louis, Missouri, USA) in PBS, pH 7.4, containing 0.01% (vol/vol) H2O2, washed again, counterstained with haematoxylin for one minute, dehydrated, cleared, and finally mounted.
Colocalisation of IgM, C3d, C5b–9, and CRP, in addition to the relative staining intensities of IgM and CRP were evaluated in each patient. Furthermore, the percentage of positive surface area was determined, by subdividing the total area of the slide into four equal parts and then estimating the percentage of positive area in each visual field. Finally, an average percentage of positive surface area was calculated from these subdivisions. The slides stained with the polyclonal antibody against IgM, or with mAbs C3-15, 5G4, or aE11 were serial slides.
Two investigators (PAJK and HWMN) assessed the anatomical localisation of the specific antibodies, as visualised by immunohistochemical staining. For the final scoring results, consensus was achieved by the two investigators.
Statistics
Statistical analysis was performed with the SPSS statistical program (Windows version 9.0). To evaluate whether observed differences were significant, paired or non-paired t tests were used when appropriate. A p value (two sided) of less than 0.05 was considered to represent a significant difference.
RESULTS
IgM deposits in the human heart
Immunohistochemical deposits of IgM were found on cardiomyocytes that were morphologically characterised as jeopardised (fig 1). IgM deposits were found on the plasma membrane and were strikingly intense in different areas of the macroscopic infarction zone (fig 1A–C), on cross striations (fig 1A), and in the cytoplasm (fig 1A) of cardiomyocytes. We used an IgM subtype antibody against Leu7 (which is not present in cardiomyocytes of the left ventricle of the adult heart) as a negative control, and the results were negative (fig 1G).
Figure 1.
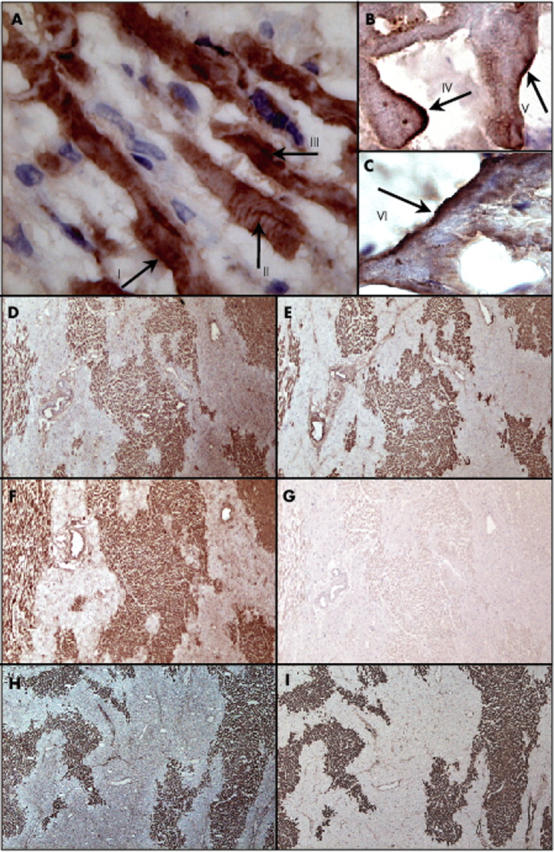
Localisation of IgM on cardiomyocytes in myocardial infarct and colocalisation of IgM, C3d, C5b–9, and C reactive protein (CRP) in the human heart. Localisation of IgM on cardiomyocytes in the heart of a patient who had died from acute myocardial infarction (AMI): (A) IgM deposited on the plasma membrane (arrow I), on cross striations (arrow II), and in the cytoplasm (arrow III) of jeopardised cardiomyocytes (original magnification, ×630); (B, C) high power view of IgM staining of the plasma membrane of jeopardised cardiomyocytes (arrows IV, V, and VI) (original magnification, ×1000). (D, H) Localisation of IgM; (E) localisation of complement factor C3d; (F) localisation of CRP; (I) localisation of complement factor C5b–9 in the heart of a patient who died after AMI (original magnification, ×100). (G) Negative staining using an IgM subtype monoclonal antibody against Leu7 (negative control).
Focal IgM deposits were found inconsistently on the endothelium of blood vessels in the heart of patients who died of AMI (not shown). This endothelial IgM staining was independent of the phase of infarction, because within each phase a subgroup of patients had no IgM staining of the endothelium, whereas others had varying amounts of IgM positive vessels. Moreover, endothelial staining for IgM was not limited to the infarction area, but also occurred at adjacent sites and remote areas of the healthy part of the heart.
IgM deposits on cardiomyocytes were found in the infarcted myocardium of patients with PMN phase infarcts and PMN phase reinfarcts. No depositions of IgM were found in the infarcted myocardium of patients with early or chronic phase infarcts or early reinfarcts. IgM deposits were never found in the healthy, remote myocardium. In addition, no IgM was found on the cardiomyocytes of a heart from a patient who died of non-heart disease related causes. As discussed earlier, no staining with IgG1 or IgG2a was found on cardiomyocytes. The replacement of specific antibody with IgG1 or IgG2a isotype controls yielded negative results in the cardiomyocytes.
Colocalisation of IgM, complement, and CRP
To test for the putative colocalisation of IgM, complement, and CRP in the infarcted myocardium, we stained serial slides of the tissue specimens. IgM (fig 1D, H) colocalised with complement factor C3d (fig 1E), complement factor C5b–9 (fig 1I), and CRP (fig 1F)—the staining patterns of IgM, CRP, and complement were strikingly similar. As was found for IgM, no complement or CRP deposits occurred at sites remote from the infarction area.
We also determined the extent of the deposits by estimating the mean surface area occupied by cardiomyocytes that were positive for IgM, complement, or CRP as a percentage of the total surface area of the slides in the infarcted region (fig 2). The IgM/complement/CRP positive surface area in patients with PMN phase infarcts or PMN phase reinfarcts was significantly greater than that of patients with early phase infarcts (p = 0.001) or early phase reinfarcts (p < 0.02). Moreover, this IgM/complement/CRP positive area tended to be greater in PMN phase reinfarcts than in PMN phase infarcts (p = 0.54 for IgM, p = 0.58 for complement; p = 0.70 for CRP). To analyse the correlation between deposits of IgM, CRP, and complement, we designed scatter plots in which the IgM/complement/CRP positive surface area of each patient was plotted, irrespective of infarct phase. This analysis revealed a linear relation between the sizes of the IgM and complement deposits (fig 3A; R = 0.999; p = 0.000), those of IgM and CRP (fig 3B; R = 0.994; p = 0.000), and those of complement and CRP (fig 3C; R = 0.996; p = 0.000).
Figure 2.

Extent of IgM, complement, and C reactive protein (CRP) deposition in the infarcted myocardium. Box plot presentation of the percentage of IgM (grey bars), complement (white bars), and CRP (shaded bars) positive myocardium. For each patient, the percentage of positive surface area for the particular antibody in relation to the total area of the examined tissue was calculated. The error bars represent minimum and maximum values, whereas the boxes represent the lower and upper quartiles. The black lines within the boxes represent the medians (N, the number of patients examined). PMN, polymorphonuclear leucocyte.
Figure 3.
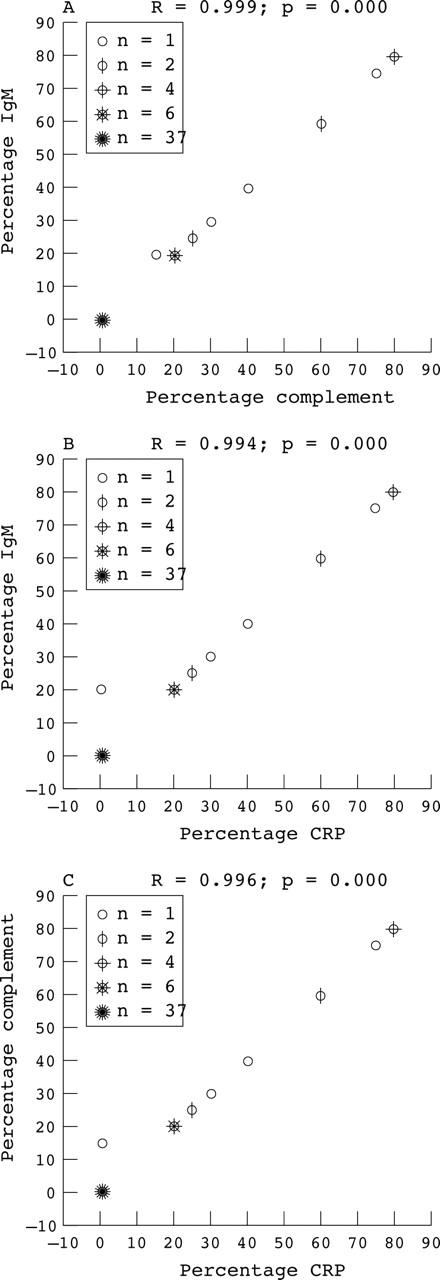
Scatter plots of the extent of IgM/complement/C reactive protein (CRP) positive areas in individual patients. Scatter plots in which the extent of deposition (expressed as per cent of surface) of IgM, complement, and CRP for each patient are plotted against each other. (A) IgM versus complement; (B) IgM versus CRP; (C) CRP versus complement. For each plot, the corresponding correlation coefficient (R) and the two tailed significance (p value) are given.
Staining intensity of IgM versus CRP
Because both IgM and CRP can activate complement, we attempted to assess the contribution of either adaptor molecule in complement activation by comparing the relative intensities of staining (fig 4). For this, only PMN phase infarcts and PMN reinfarcts were analysed because only these had IgM and CRP deposits. The staining intensity of IgM and CRP clearly varied between different patients (not shown). In the 11 examined patients with PMN phase infarcts, four patients had almost identical IgM and CRP staining intensity (IgM = CRP). In another four patients, IgM staining was less intense than that of CRP (IgM < CRP), whereas in three patients IgM staining was more intense than that of CRP (IgM > CRP). In three of the seven examined patients with PMN phase reinfarctions, IgM and CRP staining intensities were comparable, in one IgM staining was less intense than that of CRP, and in three patients IgM stained more intensely than CRP.
Figure 4.
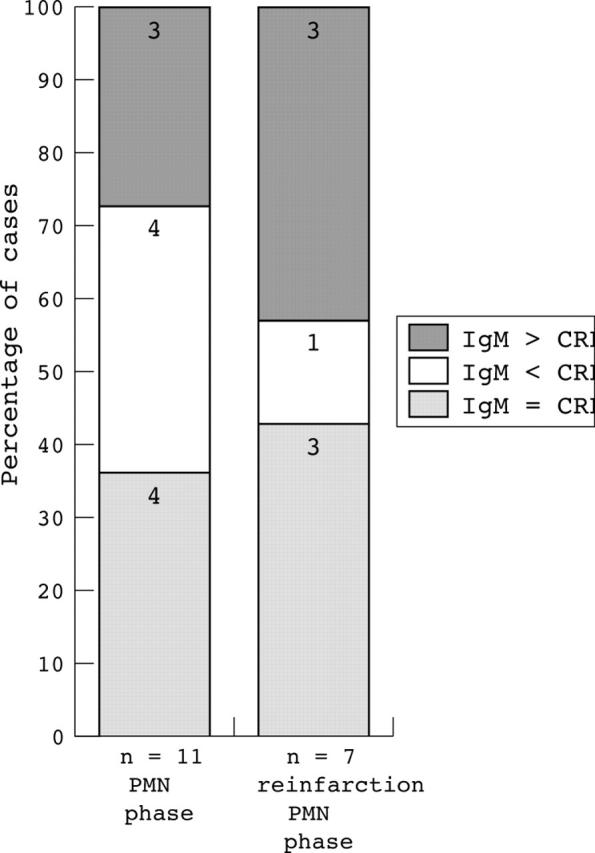
Intensity of IgM versus C reactive protein (CRP) deposition in infarcted areas of the heart. Patients were divided into one of three groups according to staining intensity: IgM staining intensity comparable to that of CRP (IgM = CRP, shaded bars), IgM staining less intense than that of CRP (IgM < CRP, white bars), and IgM staining more intense than that of CRP (IgM > CRP, grey bars). The numbers of patients in each group are given within the bars. PMN, polymorphonuclear leucocyte.
DISCUSSION
The observation that complement is locally activated by infarcted myocardium goes back over 30 years.16 Since then, it has become apparent that the complement system is an important mediator of I/R injury in the heart during AMI, and also in other organs. Studies on the molecular mechanisms of I/R induced activation of complement may offer new clues for treatment. Recent evidence shows that CRP is a complement activator in human myocardial infarcts.11 Here, we also provide evidence for the involvement of IgM in this activation. IgM is a known activator of complement,17–21 and appeared to be deposited in the infarcted human myocardium. In contrast to Schafer et al,22 who found only weak and irregular staining of IgM in the infarcted human myocardium, we found complete colocalisation of IgM with activated C3d and C5b–9, indicative of membrane attack complex formation. Irrelevant antibody controls indicated the specificity of the staining for IgM, and IgG staining yielded negative results. To our knowledge, this is the first study showing the involvement of IgM in ischaemic injury in humans.
The deposition of IgM showed a remarkable colocalisation with that of complement and CRP, strongly suggesting that the deposited IgM had bound to the same ligands in the ischaemic heart as CRP. It appears that these ligands are only exposed during the PMN phase of (re)infarction, because deposition of CRP and IgM was rarely seen during the other phases. Currently, we can only speculate about the nature of these ligands. However, microscopic evaluation revealed that the plasma membrane of the ischaemic cardiomyocytes in particular harboured the ligands for IgM and CRP, because these proteins bound to the plasma membrane of cardiomyocytes in the infarcted zone. In an earlier study, we showed that apolipoprotein H, which binds to phosphatidylserine in flip flopped membranes, colocalises with CRP in ischaemic myocardium.23 Loss of plasma membrane integrity is a feature of jeopardised cells,24 so that structures exposed in the flip flopped membrane of jeopardised cardiomyocytes might serve as ligands for CRP and IgM. Phosphorylcholine is a good candidate for such a structure because CRP is known to bind to phosphatidylcholine and particularly to lyso-phosphatidylcholine via this chemical group.4,6,25 Indeed, a considerable amount of lyso-phospholipid is generated in the infarcted myocardium.26 In addition, we have recently revealed that type II secretory phospholipase A2 (PLA2), which generates lyso-phospholipids, enhances the binding of CRP to the plasma membrane of ischaemically challenged rat cardiomyoblasts.27 Furthermore, we have found that secretory PLA2 colocalises in the infarcted human myocardium with complement and CRP,28 and therefore also with IgM. A similar specificity (preferential binding to lyso-phosphatidylcholine) has recently be described for anti-phosphorylcholine IgM.29 This IgM failed to recognise phosphatidyl lipids but did bind to lyso-phosphatidylcholine on murine apoptotic T cells, and this binding was dependent on calcium independent PLA2 activity.29 It was also revealed that increased IgM binding to the apoptotic cells was accompanied by complement activation.29 In addition to lyso-phospholipids, oxidised phospholipids in membranes may also expose phosphorylcholine in a way that allows the binding of IgM and CRP,30,31 and increased amounts of oxygen radicals are known to be generated in myocardial ischaemia.32 Its supposed specificity for phosphorylcholine is consistent with the idea that this IgM is natural IgM. Taken together, a mechanism emerges in which increased production of oxygen radicals together with enhanced PLA2 activity generates binding sites in the membrane of cardiomyocytes in the ischaemic myocardium, which promotes the binding of both natural IgM and CRP, and which ultimately leads to activation of complement and subsequent irreversible injury to the tissue.
We found differences in the staining intensity of CRP and IgM between patients, suggesting that IgM and CRP recognise and even compete for the same epitopes on the membranes of ischaemic cardiomyocytes. Serum levels of natural IgM vary between people.33 In addition, there are variations in the magnitude of the CRP response among patients suffering from AMI.10 Unfortunately, we did not have the opportunity to analyse the blood samples of the patients included in our study. Hence, the relation between circulating IgM and CRP values and the relative contribution of either protein to complement activation in the ischaemic myocardium remains to be established in further studies.
Autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and mixed connective tissue disease are accompanied by raised concentrations of IgM in the circulation,34,35 and an increased risk for cardiovascular events.36,37 In particular, increased concentrations of IgM autoantibodies against cardiolipin are thought to be the link between cardiovascular events and IgM in these diseases, particularly because patients with ischaemic heart disease may also have raised concentrations of these antibodies.38,39 Our findings could mean that in patients with autoimmune diseases, during AMI, higher IgM serum values result in greater IgM deposition in the ischaemic myocardium, thereby contributing to more extensive injury and a higher mortality. We included only one patient with autoimmune disease (scleroderma) in our study. Although this patient did have very intense staining for IgM in the infarcted myocardium, the causal link between increased concentrations of anti-phospholipid IgM autoantibodies and cardiovascular events in autoimmune disorders needs to be studied further.
Take home messages.
Both C reactive protein (CRP) and IgM target complement locally to jeopardised cardiomyocytes in the human heart after acute myocardial infarction
Localisation patterns and relative staining intensities suggest that IgM and CRP recognise similar epitopes in the ischaemic heart, but that the relative contribution of each protein to complement activation in the ischaemic myocardium differs among patients
Several studies have shown surprising similarities between CRP and IgM in cardiovascular disease. CRP has been shown to localise in ischaemic myocardium and to enhance I/R injury in the heart in a complement dependent manner.9,10 As we have shown here, IgM also localises in ischaemic myocardium, and in mouse models enhances I/R injury.12,13 CRP and IgM both enhance foam cell formation by interacting with lipoprotein particles.40,41 Furthermore, anticardiolipin IgM concentrations constitute a risk factor for atherosclerotic vascular disease,42 similar to CRP.
The role of natural IgM in vascular disease is not clear,43 and might be considered as two sides of a coin: on the one hand natural IgM antibodies against oxidised LDL protect against atherosclerosis, as was found in mice,30 rabbits44 and in humans,45 supporting the recent view that the induction of a humoral immune response to oxidised neoepitopes may be beneficial.46 On the other hand, our results suggest that IgM antibodies against epitopes in the membranes of ischaemic cardiomyocytes may enhance cardiac injury during infarction and constitute an increased risk for cardiovascular disease. Future studies should reveal the relative contribution of the effects of IgM on cardiovascular disease in humans.
Acknowledgments
Dr Niessen is a recipient of the Dr E Dekker programme of the Netherlands Heart Foundation (D99025).
Abbreviations
AMI, acute myocardial infarction
BSA, bovine serum albumin
CRP, C reactive protein
I/R, ischaemia/reperfusion
mAb, monoclonal antibody
PBS, phosphate buffered saline
PLA2, phospholipase A2
PMN, polymorphonuclear leucocyte
REFERENCES
- 1.Entman ML, Michael L, Rossen RD, et al. Inflammation in the course of early myocardial ischemia. FASEB J 1991;5:2529–37. [DOI] [PubMed] [Google Scholar]
- 2.Weisman HF, Bartow T, Leppo MK, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990;249:146–51. [DOI] [PubMed] [Google Scholar]
- 3.de Zwaan C, Kleine AH, Diris JH, et al. Continuous 48-h C1-inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J 2002;23:1670–7. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan MH, Volanakis JE. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J Immunol 1974;112:2135–47. [PubMed] [Google Scholar]
- 5.Volanakis JE, Kaplan MH. Interaction of C-reactive protein complexes with the complement system. II. Consumption of guinea pig complement by CRP complexes: requirement for human C1q, J Immunol 1974;113:9–17. [PubMed] [Google Scholar]
- 6.Volanakis JE. Complement activation by C-reactive protein complexes. Ann N Y Acad Sci 1982;389:235–50. [DOI] [PubMed] [Google Scholar]
- 7.Kushner I, Kaplan MH. Studies of acute-phase protein, I: an immunohistochemical method for the localization of Cx-reactive protein in rabbits: association with necrosis in local inflammatory response. J Exp Med 1961;114:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kushner I, Rakita I, Kaplan MH. Studies of acute phase protein, II: localization of Cx-reactive protein in heart in induced myocardial infarction in rabbits. J Clin Invest 1963;42:286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griselli M, Herbert J, Hutchinson WL, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med 1999;190:1733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagrand WK, Niessen HW, Wolbink GJ, et al. C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation 1997;95:97–103. [DOI] [PubMed] [Google Scholar]
- 11.Nijmeijer R, Lagrand WK, Lubbers YT, et al. C-reactive protein activates complement in infarcted human myocardium. Am J Pathol 2003;163:269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiser MR, Williams JP, Moore FD Jr, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med 1996;183:2343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JP, Pechet TT, Weiser MR, et al. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol 1999;86:938–42. [DOI] [PubMed] [Google Scholar]
- 14.Cotran SC, Kumar V, Robbins LR. The heart. In: Robbins pathologic basis of disease. 4th ed. Philadelphia PA: WB Saunders, 1989:605–14.
- 15.Mallory GK, White PD, Salcedo-Salgar J. The speed of healing of myocardial infarction. Am Heart J 1939;18:647–51. [Google Scholar]
- 16.Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med 1971;133:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjornson AB, Detmers PA. The pentameric structure of IgM is necessary to enhance opsonization of Bacteroides thetaiotaomicron and Bacteroides fragilis via the alternative complement pathway. Microb Pathog 1995;19:117–28. [DOI] [PubMed] [Google Scholar]
- 18.Mold C, Rodic-Polic B, Du Clos TW. Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fc gamma receptors. J Immunol 2002;168:6375–81. [DOI] [PubMed] [Google Scholar]
- 19.Parker W, Bruno D, Platt JL. Xenoreactive natural antibodies in the world of natural antibodies: typical or unique? Transplant Immunol 1995;3:181–91. [DOI] [PubMed] [Google Scholar]
- 20.Schlesinger LS, Horwitz MA. A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect Immun 1994;62:280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu X, Okada N, Iwamori M, et al. IgM natural antibody against an asialo-oligosaccharide, gangliotetraose (Gg4), sensitizes HIV-I infected cells for cytolysis by homologous complement. Int Immunol 1996;8:153–8. [DOI] [PubMed] [Google Scholar]
- 22.Schafer H, Mathey D, Hugo F, et al. Deposition of the terminal C5b–9 complement complex in infarcted areas of human myocardium. J Immunol 1986;137:1945–9. [PubMed] [Google Scholar]
- 23.Niessen HW, Lagrand WK, Rensink HJ, et al. Apolipoprotein H, a new mediator in the inflammatory changes ensuing in jeopardised human myocardium. J Clin Pathol 2000;53:863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hack CE, Wolbink GJ, Schalkwijk C, et al. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol Today 1997;18:111–15. [DOI] [PubMed] [Google Scholar]
- 25.Volanakis JE, Narkates AJ. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers and complement. J Immunol 1981;126:1820–5. [PubMed] [Google Scholar]
- 26.Van der Vusse GJ, Van Bilsen M, Reneman RS. Ischemia and reperfusion induced alterations in membrane phospholipids: an overview. Ann N Y Acad Sci 1994;723:1–14. [PubMed] [Google Scholar]
- 27.Nijmeijer R, Willemsen M, Meijer CJ, et al. Type II secretory phospholipase A2 binds to ischemic flip-flopped cardiomyocytes and subsequently induces cell death. Am J Physiol Heart Circ Physiol 2003;285:H2218–24. [DOI] [PubMed] [Google Scholar]
- 28.Nijmeijer R, Lagrand WK, Baidoshvili A, et al. Secretory type II phospholipase A(2) binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc Res 2002;53:138–46. [DOI] [PubMed] [Google Scholar]
- 29.Kim SJ, Gershov D, Ma X, et al. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med 2002;196:655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binder CJ, Horkko S, Dewan A, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 2003;9:736–43. [DOI] [PubMed] [Google Scholar]
- 31.Chang MK, Binder CJ, Torzewski M, et al. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci U S A 2002;99:13043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berges A, Van Nassauw L, Bosmans J, et al. Role of nitric oxide and oxidative stress in ischaemic myocardial injury and preconditioning. Acta Cardiol 2003;58:119–32. [DOI] [PubMed] [Google Scholar]
- 33.Stoica G, Macarie E, Michiu V, et al. Biologic variation of human immunoglobulin concentration. I. Sex–age specific effects on serum levels of IgG, IgA, IgM and IgD. Med Interne 1980;18:323–32. [PubMed] [Google Scholar]
- 34.Bakri HA, Ronnelid J, Gunnarsson I, et al. Increased serum levels of immunoglobulins, C-reactive protein, type 1 and type 2 cytokines in patients with mixed connective tissue disease. J Autoimmun 1998;11:503–8. [DOI] [PubMed] [Google Scholar]
- 35.Kumar R, Singh LM, Vaidya MP. Immunoregulatory role of stress mediators in rheumatoid arthritis. Z Rheumatol 1981;40:122–5. [PubMed] [Google Scholar]
- 36.Mutru O, Laakso M, Isomaki H, et al. Cardiovascular mortality in patients with rheumatoid arthritis. Cardiology 1989;76:71–7. [DOI] [PubMed] [Google Scholar]
- 37.Myllykangas-Luosujarvi R, Aho K, Kautiainen H, et al. Cardiovascular mortality in women with rheumatoid arthritis. J Rheumatol 1995;22:1065–7. [PubMed] [Google Scholar]
- 38.Cvetkovic JT, Wallberg-Jonsson S, Ahmed E, et al. Increased levels of autoantibodies against copper-oxidized low density lipoprotein, malondialdehyde-modified low density lipoprotein and cardiolipin in patients with rheumatoid arthritis. Rheumatology (Oxford) 2002;41:988–95. [DOI] [PubMed] [Google Scholar]
- 39.Klemp P, Cooper RC, Strauss FJ, et al. Anti-cardiolipin antibodies in ischaemic heart disease. Clin Exp Immunol 1988;74:254–7. [PMC free article] [PubMed] [Google Scholar]
- 40.Dressman J, Kincer J, Matveev SV, et al. HIV protease inhibitors promote atherosclerotic lesion formation independent of dyslipidemia by increasing CD36-dependent cholesteryl ester accumulation in macrophages. J Clin Invest 2003;111:389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu T, Borensztajn J. Macrophage uptake of low-density lipoprotein bound to aggregated C-reactive protein: possible mechanism of foam-cell formation in atherosclerotic lesions. Biochem J 2002;366:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glueck CJ, Lang JE, Tracy T, et al. Evidence that anticardiolipin antibodies are independent risk factors for atherosclerotic vascular disease. Am J Cardiol 1999;83:1490–4, A8. [DOI] [PubMed] [Google Scholar]
- 43.Rose N, Afanasyeva M. Autoimmunity: busting the atherosclerotic plaque. Nat Med 2003;9:641–2. [DOI] [PubMed] [Google Scholar]
- 44.Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci U S A 1995;92:821–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karvonen J, Paivansalo M, Kesaniemi YA, et al. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation 2003;108:2107–12. [DOI] [PubMed] [Google Scholar]
- 46.Hansson GK. Vaccination against atherosclerosis: science or fiction? Circulation 2002;106:1599–601. [DOI] [PubMed] [Google Scholar]