

Abstract
Elongin is a heterotrimeric transcription elongation factor composed of subunits A, B, and C in mammals. Elongin A and C are F-box-containing and SKP1 homologue proteins, respectively, and are therefore of interest for their potential roles in cell cycle-dependent proteolysis. Mammalian elongin C interacts with both elongin A and elongin B, as well as with the von Hippel–Lindau tumor suppressor protein VHL. To investigate the corresponding interactions in yeast, we have utilized NMR spectroscopy combined with ultracentrifugal sedimentation experiments to examine complexes of yeast elongin C (Elc1) with yeast elongin A (Ela1) and two peptides from homologous regions of Ela1 and human VHL. Elc1 alone is a homotetramer composed of subunits with a structured N-terminal region and a dynamically unstable C-terminal region. Binding of a peptide fragment of the Elc1-interaction domain of Ela1 or with a homologous peptide from VHL promotes folding of the C-terminal region of Elc1 into two regular helical structures and dissociates Elc1 into homodimers. Moreover, analysis of the complex of Elc1 with the full Elc1-interaction domain of Ela1 reveals that the Elc1 homodimer is dissociated to preferentially form an Ela1/Elc1 heterodimer. Thus, elongin C is found to oligomerize in solution and to undergo significant structural rearrangements upon binding of two different partner proteins. These results suggest a structural basis for the interaction of an F-box-containing protein with a SKP1 homologue and the modulation of this interaction by the tumor suppressor VHL.
The elongin (SIII) complex strongly stimulates the rate of elongation by RNA polymerase II by suppressing transient pausing by polymerase at many sites along the DNA (1, 2). In mammals, elongin is a trimeric complex. Elongin A is the catalytic subunit. Elongins B and C form a binary complex that is capable of enhancing the transcriptional activity of elongin A (3, 4). Elongin C binds directly to elongin A in the absence of elongin B to form an elongin A/C complex with increased specific activity in transcription. In contrast, elongin B does not bind stably to elongin A in the absence of elongin C. Instead, elongin B appears to play a chaperone-like role in the assembly of the elongin A/B/C complex by binding to elongin C and promoting its interaction with elongin A.
The product of the von Hippel–Lindau (VHL) tumor suppressor gene also binds tightly and specifically to the elongin B/C complex in vitro (5) and in vivo (6). When the VHL protein binds to elongin B/C, it inhibits the ability of elongin B/C to interact with elongin A. Binding of VHL and elongin A to the elongin B/C complex in vitro is mutually exclusive and depends on a short sequence conserved in VHL and elongin A (3, 6, 7). This region of VHL is frequently mutated in patients with the VHL disease, a genetic cancer syndrome characterized by the development of multiple tumors, including renal carcinomas, retinal angiomas, cerebellar hemangioblastomas, and pheochromocytomas (8–11). These mutations cause VHL to lose its ability to interact with elongin B/C. The elongin B/C subcomplex has also been shown to interact with SOCS box-containing proteins (12). These protein–protein interactions appear to be mediated by the elongin C subunit.
The functional domains of mammalian (rat) elongin C have been characterized by mutagenesis studies (13). An N-terminal region (Tyr-18 to Ile-30; see Fig. 1) is important for binding to mammalian elongin B, formation of the A/B/C complex, and activation of elongin A. Mutations in the extreme C terminus (Glu-98 to Cys-112) of elongin C have dramatic effects on elongin A/B/C formation but not on elongin B binding. Activation of elongin A is also affected by mutations in residues Asn-61 to Pro-97. The molecular mechanisms by which these residues contribute to the overall function of the elongin C complex is not known.
Figure 1.

Amino acid sequences and alignment of yeast Elc1 (Yeast C) with rat elongin C (Rat C) and yeast Skp1 (Yeast Skp1). (A) blast alignment of rat elongin C and yeast Elc1. Identical residues are indicated by a colon. Similar residues are indicated by a period. The symbols [ ], ( ), and { } indicate the sites in rat elongin C that were previously reported to be involved in elongin B binding, elongin A/B/C complex formation, and elongin A activation, respectively (13). (B) Alignment of yeast Skp1 with yeast Elc1. Identical residues are indicated by a colon. Similar residues are indicated by a period. The yeast Elc1 construct used in our studies has an additional three residues, GSH, at the N terminus for a total of 102 residues.
Yeast homologues of mammalian elongin A (Ela1) and C (Elc1) have been identified on the basis of sequence similarity (14, 15). There is no obvious elongin B homologue in the yeast genome. The yeast C homologue exhibits 41% identity and 71% similarity over a 91-residue region of mammalian elongin C (15). The greatest similarity to mammalian elongin C spans residues Glu-92 to Cys-112, which includes a region that binds to VHL and activates human elongin A. Residues 1–143 of Ela1 display 31% identity to mammalian elongin A; the greatest similarity is in the region of the mammalian protein most critical for transcriptional activity (7). We have shown that Ela1 and Elc1 form a stable complex (14), which serves as further evidence that these two proteins are the yeast homologues of mammalian elongin A and C.
The tripartite complex of mammalian elongin A, elongin B, and elongin C shares sequence and structural similarities with the SCF complexes [Skp1–Cdc53 (cullin)–F-box protein] involved in cell cycle- and ubiquitin-dependent proteolysis (16–19). Elongin A contains an F-box motif and elongin B, a ubiquitin-like domain (17, 20). Elongin C is a SKP1 homologue in which one region of high similarity corresponds to the elongin B binding site (Fig. 1) (13, 20). While the analogy between elongin and the SCF complex is striking, to date there is no direct evidence that elongin participates in the regulation of cell cycle-dependent proteolysis. Here we present biochemical and structural data for yeast Elc1 that strengthens the analogy between Elc1 and SKP1. We show that, like SKP1, Elc1 is a homodimer in the absence of its F-box partner (Ela1), but preferentially forms a heterodimer in the presence of residues 1–143 of Ela1 [Ela1(1–143)]. Using NMR spectroscopy, we show that both Ela1 and a VHL peptide bind to a common C-terminal region of Elc1, which in the absence of a binding partner contains dynamically unstable α-helical structure.
MATERIALS AND METHODS
Sample Preparation.
Procedures for Escherichia coli expression and purification of unlabeled, uniformly 15N-, and 15N,13C-labeled Elc1 [free and bound to unlabeled Ela1(1–143)] are described by Koth et al. (14). Final NMR sample conditions for free Elc1 and Ela1(1–143)/Elc1 complex were 10 mM sodium phosphate (NaPi), pH 7.5, 500 mM NaCl, 10 μM ZnSO4, 100 μM EDTA, 7.5 mM DTT, and 0.7–1.5 mM protein concentration. Complexes of Elc1 (15N- and 15N,13C-labeled) and unlabeled VHL peptide were generated by titration of 61-μl aliquots of an 18-mg/ml solution of VHL(157–171) (NH2-TLKERCLQVVRSLVK-CO2H) into a 1.5 mM Elc1 NMR sample. Similarly, 51-μl aliquots of a 10-mg/ml solution of unlabeled elongin A peptide (NH2-SLQTLCEISLMRNHS-CO2H), referred to here as Ela1(3–17), was added to 0.7 mM 15N-labeled Elc1 to produce Ela1(3–17)/Elc1 complex. The buffer for these two complexes was as described above except for a lower NaCl concentration (100 mM) and pH (7.0).
NMR Spectroscopy and Resonance Assignments.
NMR experiments were performed on Varian Unity and Unity+ and Bruker AMX and DRX spectrometers operating at 500-, 600-, and 800-MHz proton frequencies. NMR experiments with VHL(157–171)/Elc1 and Ela1(3–17)/Elc1 were run at 30°C. All other experiments were carried out at 25°C. 15N heteronuclear single quantum correlation (HSQC) (21) experiments were obtained for 15N-labeled free and bound Elc1 samples. Sequence-specific resonance assignments were made for 15N,13C-labeled free Elc1 and Elc1 complexed to VHL(157–171) by using the following triple-resonance experiments: HNCO (22), CBCANH (23), CBCA(CO)NH (24), CC(CO)NH-TOCSY (25), HBHA(CBCACO)NH (26), and HCC(CO)NH-TOCSY (25) [TOCSY, total correlation spectroscopy; NOESY, nuclear Overhauser effect (NOE) spectroscopy]. For 15N- and 15N,13C-labeled free and VHL(157–171)-bound Elc1, three-dimensional 15N NOESY-HSQC, 15N TOCSY-HSQC, 13C NOESY-HSQC, 13C HCCH-TOCSY, and 13C HCCH-COSY (27–30) were acquired to confirm and extend sequential assignments. Mixing times of 80 and 100 ms were used for the three-dimensional NOESY experiments. An additional 15N HSQC was obtained for free Elc1 prepared in the same buffer as VHL(157–171)-bound Elc1, at 30°C.
High-resolution homonuclear two-dimensional (2D) NOESY (31), TOCSY (32), and 2Q (33) experiments were performed on unlabeled free and VHL(157–171)-bound Elc1 samples dissolved in 10% D2O/90% H2O and in 100% D2O (D2O = 2H2O). Mixing times for the 2D NOESY experiments were 30 and 100 ms. A 300-ms 2D NOESY spectrum was acquired for free VHL(157–171) at 10°C and an 80-ms 2D 15N-filtered-NOESY spectrum (34) at 30°C was acquired for VHL(157–171) bound to 15N-labeled Elc1. A three-dimensional 13C filter-edited NOESY spectrum (35) with a mixing time of 150 ms was run at 30°C, to detect intermolecular NOEs between VHL(157–171) and 15N,13C-labeled Elc1.
Data were processed and analyzed with the NMRPipe/NMRDraw software (36) and analyzed with the Pipp program (37) and NMRView analysis package (38) on Sun SPARC5 and SPARC20 workstations. Typically, Lorentzian-to-Gaussian or shifted sine windows were applied before Fourier transformations. Data were usually zero filled in all three dimensions. Where applicable, linear prediction was also employed. The proton chemical shifts were referenced to the 0 ppm methyl resonance of external 2,2-dimethylsilapentane-5-sulfonic acid (DSS) dissolved in the appropriate buffer (39). Nitrogen and carbon chemical shifts were indirectly referenced using ratios of 0.101329118 and 0.25144953, respectively (40). Chemical shift indices (CSIs) of 1H and 13C nuclei were calculated with the csi program (41).
Analytical Ultracentrifugation.
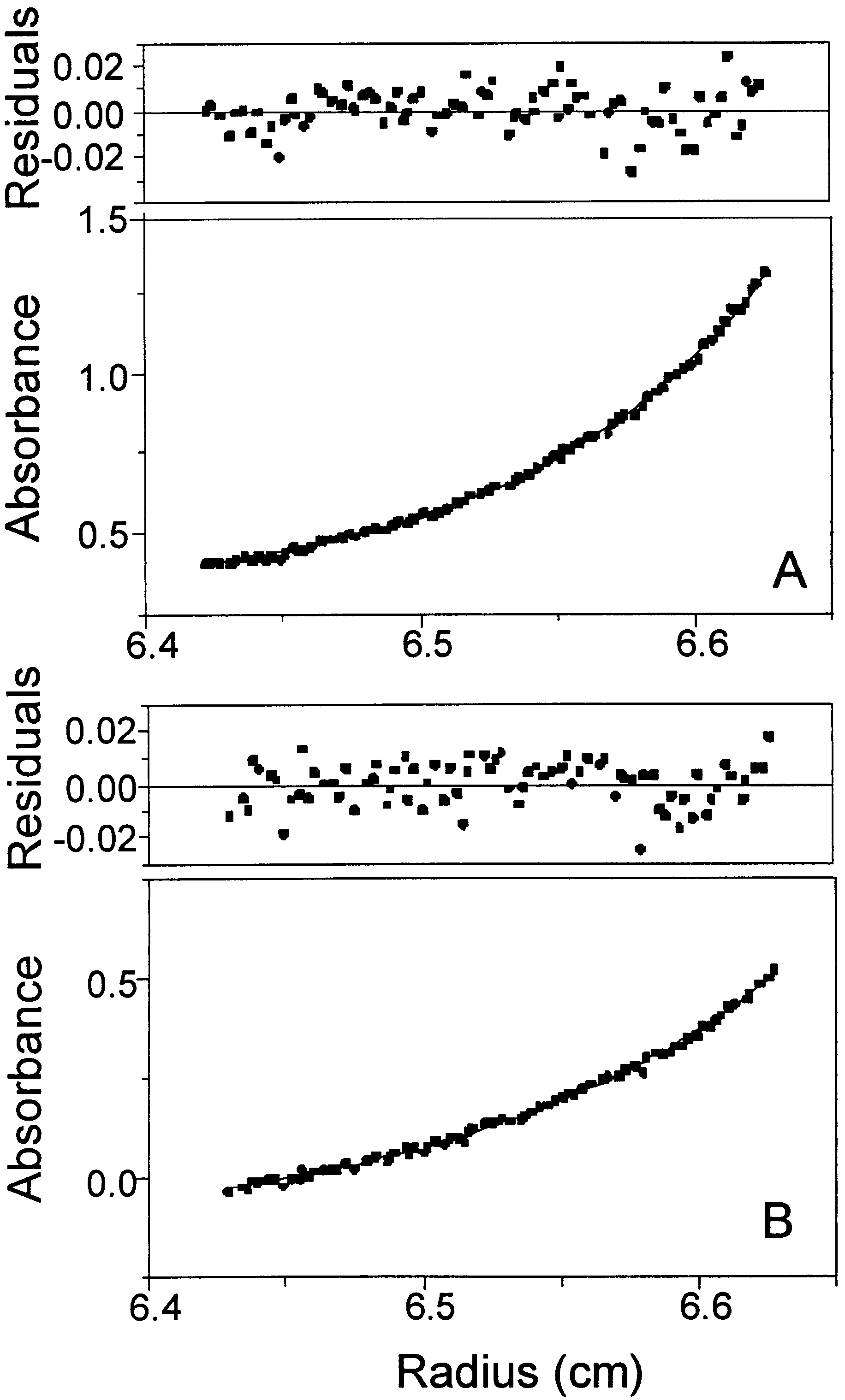
Sedimentation experiments were performed at 20°C on a Beckman XLI analytical ultracentrifuge with an AN50-Ti rotor. The sedimentation equilibrium runs using six-channel charcoal–Epon cells were performed for approximately 34 hr before equilibrium absorbance measurements were taken. Molecular weight determinations involved global analysis of three sample concentrations (0.1, 0.3, and 0.7 mg/ml) centrifuged at three different speeds (16,000, 17,500, and 19,000 rpm). Molecular weights were calculated with Beckman XLI data analysis software in which absorbance vs. radial position data were fitted to the sedimentation equilibrium equation by using nonlinear least-squares techniques (42). The partial specific volume and density of the sample were calculated by using the program Sedinterp (43) from the amino acid sequence and buffer composition, respectively.
Sedimentation velocity measurements using double-sector charcoal–Epon cells equipped with sapphire windows were performed on samples (4 mg/ml) centrifuged at 40,000 rpm, and concentration distributions were determined by using Raleigh interference optics. Time derivative analysis (44) of the sedimentation velocity data was performed with the Beckman XLI data analysis software.
RESULTS
Yeast Elc1 Forms Homo-oligomers and Has Regions of Dynamic Instability.
In an effort to characterize the structure of elongin C and corresponding protein–protein complexes, we analyzed Elc1 by NMR spectroscopy. In general, the linewidths of resonance peaks were broader than expected, and many of the triple-resonance NMR spectra were of low sensitivity, suggesting Elc1 has a longer rotational correlation time than expected for a monomeric protein of ≈12 kDa. We also observed a drastic increase in the linewidths of Elc1 resonances with increasing concentration (from 0.3 mM to 1 mM), suggesting that Elc1 self-aggregates. Therefore, equilibrium sedimentation and sedimentation velocity studies (see supplemental text and Figs. 5 and 6, which are available on the PNAS web site, www.pnas.org) were performed to determine the quaternary structure of Elc1. Both of these methods indicate that Elc1 forms a single species with an apparent molecular mass the size of a tetramer (42–44 kDa, for the two methods; the expected molecular mass of a tetramer is 47 kDa). Interestingly, we were able to acquire triple-resonance NMR spectra, albeit with low sensitivity, even though these experiments are not expected to be successful for proteins the size of an Elc1 tetramer (45). The apparent contradiction between NMR and sedimentation results could possibly be explained by a model for tetramerization in which two dimers form with high affinity and these in turn form a tetramer through a flexible region of the protein. The fact that only one set of resonances is observed for each residue indicates that Elc1 forms a symmetric oligomer. In the model each dimer tumbles relatively independently in solution, resulting in a correlation time closer to that of a dimer as opposed to a tetramer. Evidence supporting this model is presented below.
Several features of the NMR spectra indicate that Elc1 has regions of dynamic instability and/or flexibility. First, only 74% of the expected peaks were observed in the 15N HSQC spectrum (Fig. 2A), indicating that a subset of residues in Elc1 have dynamic properties that broaden and obscure their NH resonances beyond the broadening caused by oligomerization. Second, the peaks that are visible in the HSQC spectrum are not of uniform intensity. Finally, a significant number (11%) of NH peaks that are visible in the HSQC spectrum do not show correlations in triple-resonance or 15N-edited NOESY spectra. These features are not consistent with a random coil, which would give rise to high-intensity peaks with narrow linewidths in all of the NMR spectra. In total, we were able to assign 64% of the backbone and Cβ resonances of Elc1. Most, but not all, of the peaks missing from the HSQC spectrum and those lacking correlations in triple-resonance and NOESY spectra are from residues at the C terminus of Elc1. Thus, the dynamic instability of Elc1 appears to be most prominent at the C terminus, but also extends to a lesser extent throughout the protein.
Figure 2.

15N HSQC spectra of 15N-labeled Elc1. (A) Free. (B) VHL(157–171)-bound. (C) Ela1(1–143)-bound. (D) Ela1(3–17)-bound. Peaks assigned are labeled. Samples A, B, and D are 0.7–1.5 mM in 10 mM NaPi, pH 7.0/100 mM NaCl/10 μM ZnSO4/100 μM EDTA/7.5 mM DTT in 90% H2O/10% D2O. Sample C is 0.7 mM in similar buffer but at pH 7.5 and 500 mM NaCl. Experiments were run at 30°C (A, B, and D) or 25°C (C).
The secondary structure of Elc1 was identified by using both CSI (Fig. 3A) and NOE data. Two β-strands joined by a short turn are found at the N terminus: strand 1, Gln-3 to Val-9, and strand 2, Asp-12 to Ile-18. These are followed by a long helical region involving residues Ser-19 to Ser-40. Beyond Ser-40, the evidence for regular secondary structural elements in the Elc1 dimer is more ambiguous. However, the data suggest the presence of an unstable β-strand and a helix in the regions Arg-43 to Ile-50 and His-52 to Gly-69, respectively.
Figure 3.

Plots of consensus CSI values for free Elc1 (A) and VHL(157–171)-bound Elc1 (B). CSI values of +1 reflect residues with helical backbone conformation, and values of −1 reflect regions with extended (β-type) backbone conformation. Unassigned residues are given the value of +0.2. Predicted helices are represented by coiled ribbons and β-strands by arrows.
The two N-terminal β-strands interact with each other in an antiparallel manner. This conclusion is based on a number of unambiguous long-range Hα–NH, Hα–Hα, and NH–NH NOEs between the two strands (Fig. 4). β-Strand 1 and Asp-13 from strand 2 also interact with residues Arg-43 to Ile-50 in the central part of Elc1, and several unambiguous long-range NOEs suggest that this latter region contributes a third parallel β-strand to the β-sheet.
Figure 4.

Schematic representation of β-strand interactions in Elc1. Inter-strand long-range NOEs are indicated by arrows.
A Mammalian VHL Peptide Binds to Yeast Elc1 and Induces Formation of Two C-Terminal α-Helices.
Elc1 interacts with mammalian elongin A and VHL. The interaction of Elc1 with these proteins is mediated in part by a homologous region found in the N-terminal regions of both elongin A and VHL (6, 7, 46). To study the interaction of Elc1 with elongin A and VHL, we performed a titration of a synthetic VHL peptide with Elc1 and monitored the interaction by NMR spectroscopy. At substoichiometric concentrations of the peptide, we observed two sets of resonances that correspond to the free Elc1 oligomer and to the VHL(157–171)/Elc1 complex. This property indicates that the Elc1/peptide complex is long-lived on the NMR time scale, with a dissociation constant Kd ≪ 1 μM and a lifetime greater than 50 ms, assuming a diffusion-limited on-rate of 108 M−1⋅s−1.
A 2D homonuclear NOESY spectrum of the VHL peptide shows that it has a random coil conformation in the absence of Elc1. However, upon addition of Elc1, a region of the peptide adopts a helical conformation. A continuous stretch of seven NH–NH NOEs were detected in the 2D 15N-filtered-NOESY spectrum of the complex. Identification of the residues in this helical region of VHL(157–171) awaits more detailed NMR analysis of this peptide in the complex with Elc1.
A 15N HSQC spectrum of Elc1 saturated with the VHL peptide is shown in Fig. 2B. In contrast to the HSQC spectrum of Elc1 alone, the spectrum of the VHL(157–171)/Elc1 complex contains almost all expected backbone amide peaks. Moreover, all four side-chain amide resonances (Gln-3, Gln-48, Asn-62, Asn-64) were observed. Thus, binding of the VHL peptide appears to have a global stabilizing effect on Elc1. A sedimentation velocity experiment on the VHL(157–171)/Elc1 complex (Fig. 6 in the supplemental data on www.pnas.org) showed that in the presence of VHL, Elc1 forms a single species the size of a dimer (apparent molecular mass 28 kDa; expected molecular mass of dimer is 23.6 kDa). We also note that during the peptide titration the NMR spectral changes occurred very slowly. The intensity of the new signals increased with time for a period as long as several hours, suggesting that the kinetics of binding is very slow, or that dissociation from tetramers to dimers is slow.
Many of the resonances from the N-terminal half of free and VHL(157–171)-bound Elc1 have very similar chemical shift patterns (compare Fig. 2 A and B). This fact, combined with the improved quality of most NMR spectra, allowed us to assign 94% of Elc1 backbone residues in the VHL(157–171)/Elc1 complex. The changes in backbone 1HN and 15N chemical shifts of assigned residues of free and VHL(157–171)-bound Elc1, measured under identical conditions, are summarized in Fig. 7 (supplemental data, www.pnas.org). The VHL peptide did not seem to significantly affect residues Met-1 to Ile-18, which constitute the N-terminal β-sheet. Residues Gln-3 to Ile-18 experience very small 15N (<0.5 ppm) and amide 1H chemical shift perturbations (≤0.1 ppm). Furthermore, similar long-range NOEs between β-strand 1 and β-strand 2 as observed for free Elc1 were also present in the three-dimensional 15N NOESY-HSQC of VHL(157–171)-bound Elc1. The largest changes in chemical shift position are for residues Ile-25, Ser-26, and several residues in the central region (Ile-50 to Ile-77). Although large chemical shift perturbations are not evident for C-terminal residues, this is because of the fact that most of the resonances for these residues were not assigned in free Elc1.
Compared with free Elc1, VHL(157–171)-bound Elc1 has additional regions of stable secondary structure, on the basis of CSI (Fig. 3B) and observed NOE patterns. These include a β-strand from residues Gly-42 to Lys-47 (or possibly Phe-49) and two helices from His-52 to Gly-69 and from Thr-84 to Tyr-96. While regular secondary structure in these regions could not be assigned in free Elc1, the data do not support random coil conformation. Thus, the appearance of these new elements of secondary structure in VHL(157–171)/Elc1 is attributed to stabilization of pre-existing but dynamically unstable structural elements. The two new helices appear to be separated by a long 13-residue loop that is both highly mobile and, to a certain degree, ordered. The 15N HSQC peaks for residues Val-70 to Ile-82 are of much greater intensity than the rest of the backbone amide signals, indicating that this region has greater mobility. A stretch of NH–NH connectivities within this loop are also observed in the 15N NOESY-HSQC spectrum of VHL(157–171)-bound Elc1, suggesting that this loop has some degree of ordered structure.
The greatest effects of the VHL peptide on Elc1 are seen in the C-terminal residues. Preliminary analysis of the 13C filter-edited NOESY spectrum of VHL(157–171)-bound Elc1 indicates that the peptide makes several contacts to side-chain protons of Elc1: γCH3 and δCH3 of Ile-77, δCH3 of Ile-82, βH and γCH3 of Thr-84, and βH of Ala-94. The above observations, combined with the fact that the peptide dissociates Elc1 from tetramers into dimers, suggest that association of Elc1 dimers into tetramers is mediated by its C-terminal residues. The extreme C terminus of elongin C is predicted by the Chou and Fasman (47) and Garnier (48) algorithms to form a short, hydrophobic α-helix that has the potential to form a coiled-coil protein–protein interaction domain. Small amphipathic peptides with sequences similar to those of residues Ile-77 to Ile-99 of Elc1 are capable of self-association into molten globule-like helical bundles (49). Given that the C terminus of Elc1 appears to mediate dimer–dimer association and has molten globule-like dynamic properties, we propose that the interface between dimers to form tetramers is conformationally heterogeneous and flexible enough to allow the N-terminal regions of Elc1 to behave similar to free dimers in many NMR experiments.
Ela1 Interacts with the C Terminus of Elc1.
Elongin A shares a region of sequence homology to VHL (3, 6, 7). To analyze the interaction of elongin A with Elc1, and to compare the structures of Elc1 in each complex, we studied Elc1 complexed with both Ela1(1–143) and Ela1(3–17).
The 2D HSQC spectrum of 15N-labeled Elc1 bound to unlabeled Ela1(1–143) (Fig. 2C) has many peaks that coincide with those of VHL(157–171)-bound Elc1 but are absent from the HSQC spectrum of free Elc1. These include Glu-55, Val-58, Ile-82, Glu-85, Leu-89, Leu-92, Ala-94, Asp-95, and Leu-97, which are distinctive resonances of helices 2 and 3 and the intervening loop. This suggests that the C-terminal part of Elc1 adopts a similar conformation in the presence of Ela1(1–143) and VHL(157–171).
Upon addition of the Ela1(3–17) peptide, Elc1 exhibits intermediate exchange on the chemical shift time scale, in contrast to the slow exchange observed for the VHL peptide, indicating that Ela1(3–17) has lower affinity for Elc1 than does the VHL peptide. Moreover, even in the presence of excess Ela1(3–17) several peaks are missing in the HSQC spectrum, namely Ile-77, Glu-81, Glu-85, Ser-87, Leu-88, and Leu-90 to Ile-99. This observation parallels the absence of resonances seen for the dynamically unstable (molten globule-like) C terminus of free Elc1, although there are fewer missing resonances for the complex with Ela1(3–17). The resonances of the remainder of the residues of Ela1(3–17)-bound Elc1 are superimposable with those of VHL(157–171)-bound Elc1 (compare Fig. 2 B and D). Thus, the C-terminal region of Elc1 is most affected by binding of Ela1(3–17), consistent with the idea that Ela1(3–17) and VHL(157–171) interact with the same region of Elc1.
Ela1 and Elc1 Form a 1:1 Heterodimer.
Our studies of free Elc1 showed that a symmetric homodimer is formed, which in turn dimerizes to form a tetramer. To identify the stoichiometry of the Ela1(1–143)/Elc1 interaction, we performed equilibrium sedimentation studies on this complex. These data demonstrate quite convincingly that Ela1(1–143)/Elc1 forms a 1:1 heterodimer with an apparent molecular mass of 31.5 kDa (expected molecular mass of dimer is 29 kDa). It is not immediately obvious how Ela1(1–143) disrupts further the oligomerization of Elc1. Comparison of the 15N HSQC spectra of VHL(157–171)-bound Elc1 (Fig. 2B) and Ela1(1–143)-bound Elc1 (Fig. 2C) reveals peaks that disappear, narrow, and/or are greatly displaced upon binding of the peptide. We find that the residues affected are not confined to specific regions of Elc1. Isotope-filtered NMR experiments should allow us to map the interaction surface of Elc1 with Ela1(1–143).
DISCUSSION
Our NMR data reveal an important role for protein dynamics in the interactions of Elc1 with its protein partners. First, binding of partner proteins or peptides to Elc1 induces and/or stabilizes two helical elements in the C terminus of Elc1. Second, the VHL peptide folds into a helical structure upon binding to Elc1. Third, our data suggest that there may be another region besides the C-terminal helix of Elc1 which may be involved in interactions with Ela1(1–143). A large interaction surface between Elc1 and Ela1(1–143) could explain the strong structural and thermodynamic stabilizing effect seen for the interaction of these two proteins (14). Using CD spectroscopy, we have shown that Ela1(1–143) is unfolded in the absence of Elc1 and that there is a large increase in helical content upon formation of the Ela1(1–143)/Elc1 heterodimer (14). This coupling of folding with Ela1(1–143)-Elc1 binding can be explained by both induction of the C-terminal helices in Elc1 and folding of Ela1(1–143).
In this report, we provide evidence supporting the analogy between elongin and SCF complexes. So far the homologies between elongin and a number of SCF complexes are largely based on sequence similarities, and there is still insufficient structural information to draw direct conclusions. Functionally, there are no data to support that elongin is a ubiquitin ligase as most of the SCF complexes are. However, recent evidence links VHL to regulation of protein stability (50). Our data support a structural similarity between Elc1 and SKP1 beyond that suggested by sequence comparison. SKP1 has recently been shown by gel filtration and gradient centrifugation experiments to be capable of forming dimers, but the residues responsible are not known (51). Our observations support formation of an Elc1 dimer, and retention of this dimer when bound to the human VHL peptide.
Since the submission of this manuscript, a paper has been published that describes the structure of the ternary complex of mammalian VHL–elongin C–elongin B (VCB) (52). VCB is a 1:1:1 heterotrimer. Elongin C consists of a three-stranded β-sheet packed against four α-helices. Interestingly, Stebbins et al. (52) noticed that the overall fold of mammalian elongin C is very similar to that of the tetramerization domain of the Shaker potassium channel (52, 53). The biological significance of this observation is strengthened by our sedimentation data showing that free Elc1 is a tetramer.
Our NMR-derived structural data for the yeast Elc1/VHL peptide complex agree with all of the major features of the crystal structure of VCB. The secondary structure and topology of Elc1 correspond to those of elongin C in the crystal structure. Moreover, the interaction with VHL appears to be quite similar. In the crystal structure, the C-terminal helices of elongin C are separated by a long, well-ordered loop in an extended conformation. This extended loop together with three of the helices form a concave surface with a central pocket into which the H1 helix of the VHL α domain fits. The interaction surface is dominated by hydrophobic residues. Our studies provide evidence that VHL(157–171), which is analogous to H1 of the VHL α domain, assumes a helical conformation upon binding Elc1 and that the interactions are also mediated mainly by hydrophobic residues.
Our findings provide useful insights into key structural properties of yeast elongin C and the corresponding mammalian proteins. As previously noted, Elc1 exhibits very high sequence homology to mammalian elongin C; Elc1 also binds mammalian elongin A and VHL proteins and can even induce elongin A’s transcriptional activity (15). Elc1 does not exhibit any affinity for human elongin B (54). Since no yeast elongin B homologue could be identified despite a complete analysis of the yeast genome, the homodimerization of Elc1 may play a role similar to that of elongin B/C heterodimerization. We postulate that C/C dimerization may fulfill the role played by B/C heterodimerization in mammalian systems.
Supplementary Material
Acknowledgments
We thank S. Go for help with sedimentation data and Drs. L. E. Kay and J. Chung for pulse programs and assistance with NMR experiments. This research was supported by the National Cancer Institute of Canada, the National Institutes of Health (U.S.), a Medical Research Council of Canada Fellowship to C.M.K., and a Human Frontier Science Program Organization Postdoctoral Fellowship to G.M.
ABBREVIATIONS
- VHL
von Hippel–Lindau
- Ela1 and Elc1
yeast homologues of elongin A and elongin C, respectively
- SCF
Skp1–Cdc53 (cullin)–F-box protein
- HSQC
heteronuclear single quantum correlation
- TOCSY
total correlation spectroscopy
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
- 2D
two-dimensional
- CSI
chemical shift index
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Bradsher J N, Jackson K W, Conaway R C, Conaway J W. J Biol Chem. 1993;268:25587–25593. [PubMed] [Google Scholar]
- 2.Bradsher J N, Tan S, McLaury H-J, Conaway J W, Conaway R C. J Biol Chem. 1993;268:25594–25603. [PubMed] [Google Scholar]
- 3.Aso T, Lane W S, Conaway J W, Conaway R C. Science. 1995;269:1439–1443. doi: 10.1126/science.7660129. [DOI] [PubMed] [Google Scholar]
- 4.Garrett K P, Aso T P, Bradsher J N, Foundling S I, Lane W S, Conaway R C, Conaway J W. Proc Natl Acad Sci USA. 1995;92:7172–7176. doi: 10.1073/pnas.92.16.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan D R, Pause A, Burgess W H, Aso T, Chen D Y, Garrett K P, Conaway R C, Conaway J W, Linehan W M, Klausner R D. Science. 1995;269:1402–1406. doi: 10.1126/science.7660122. [DOI] [PubMed] [Google Scholar]
- 6.Kibel A, Iliopoulos O, DeCaprio J A, Kaelin W G. Science. 1995;269:1444–1446. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 7.Aso T, Haque D, Barstead R J, Conaway R C, Conaway J W. EMBO J. 1996;15:5557–5566. [PMC free article] [PubMed] [Google Scholar]
- 8.McKusick V A. Mendelian Inheritance in Man. Baltimore: Johns Hopkins Univ. Press; 1992. [Google Scholar]
- 9.Latif F, Tory K, Gnarra J, Yao M, Duh F, Orcutt M L, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Science. 1993;260:1312–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 10.Gnarra J R, Tory K, Weng Y, Schmidt L, Wei M H, Li H, Latif F, Liu S, Chen F, Duh F M, et al. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 11.Kishida T, Stackhouse T M, Chen F, Lerman M I, Zbar B. Cancer Res. 1995;20:4544–4548. [PubMed] [Google Scholar]
- 12.Kamura T, Sato S, Haque D, Liu L, Kaelin W G, Conaway R C, Conaway J W. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takagi Y, Conaway R C, Conaway J W. J Biol Chem. 1996;271:25562–25568. doi: 10.1074/jbc.271.41.25562. [DOI] [PubMed] [Google Scholar]
- 14.Koth, C., Botuyan, M. V., Jansma, D., Chazin, W. J., Friesen, J. D., Arrowsmith, C. H. & Edwards, A. E. (1999) J. Biol. Chem.274, in press. [DOI] [PubMed]
- 15.Aso T, Conrad M N. Biochem Biophys Res Commun. 1997;241:334–340. doi: 10.1006/bbrc.1997.7819. [DOI] [PubMed] [Google Scholar]
- 16.Feldman R M R, Correll C C, Kaplan K B, Deshailes R J. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 17.Skowyra D, Craig K L, Tyers M, Elledge S J, Harper J W. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 18.Patton E E, Willems A R, Sa D, Kuras L, Thomas D, Craig K L, Tyers M. Genes Dev. 1998;12:692–705. doi: 10.1101/gad.12.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krek W. Curr Opin Genet Dev. 1998;8:36–42. doi: 10.1016/s0959-437x(98)80059-2. [DOI] [PubMed] [Google Scholar]
- 20.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper J W, Elledge S J. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 21.Zhang O, Kay L E, Olivier J P, Forman-Kay J D. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 22.Muhandiram D R, Kay L E. J Magn Reson Ser B. 1994;103:203–216. [Google Scholar]
- 23.Wittekind M, Mueller L. J Magn Reson Ser B. 1993;101:201–205. [Google Scholar]
- 24.Grzesiek S, Anglister J, Bax A. J Magn Reson Ser B. 1993;101:114–119. [Google Scholar]
- 25.Grzesiek S, Bax A. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 26.Grzesiek S, Bax A. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 27.Marion D, Driscoll P C, Kay L E, Wingfield P T, Bax A, Gronenborn A M, Clore M G. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 28.Kay L E, Guang-Yi X, Singer A U, Muhandiram D R, Forman-Kay J D. J Magn Reson Ser B. 1993;101:333–337. [Google Scholar]
- 29.Bax A, Clore M G, Driscoll P C, Gronenborn A M, Ikura M, Kay L E. J Magn Reson. 1990;87:620–627. [Google Scholar]
- 30.Bax A, Clore M G, Gronenborn A M. J Magn Reson. 1990;88:425–431. [Google Scholar]
- 31.Jeener J, Meier B H, Bachmann P, Ernst R. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 32.Cavanagh J, Rance M. J Magn Reson. 1992;96:670–678. [Google Scholar]
- 33.Braunschweiler L, Bodenhausen G, Ernst R R. Mol Phys. 1983;48:535–560. [Google Scholar]
- 34.Ikura M, Bax A. J Am Chem Soc. 1992;114:2433–2440. [Google Scholar]
- 35.Zwahlen C, Legault P, Vincent S J F, Greenblatt J, Konrat K, Kay L E. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Garrett D S, Powers R, Gronenborn A M, Clore G M. J Magn Reson. 1991;95:214–220. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Johnson B, Blevins R A. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 39.Wishart D S, Sykes B D. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 40.Wishart D S, Bigam C G, Yao J, Abilgaard F, Dyson H J, Oldfield E, Markley J L, Sykes B D. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 41.Wishart D S, Sykes B D, Richards F M. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 42.Johnson M L, Correia J J, Yphantis D A, Halvorson H R. Biophys J. 1981;36:575–588. doi: 10.1016/S0006-3495(81)84753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laue T M, Shah B D, Ridgeway T M, Pelletier S L. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding S, Rowe A, editors. Cambridge, U.K.: R. Soc. Chem.; 1992. pp. 90–125. [Google Scholar]
- 44.Stafford W. Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 45.Arrowsmith C H, Wu Y-S. Prog NMR Spectrosc. 1998;32:277–286. [Google Scholar]
- 46.Gnarra J R, Zhou S, Merrill M J, Wagner J R, Krumm A, Papavassiliou E, Oldfield E H, Klausner R D, Linehan W M. Proc Natl Acad Sci USA. 1996;93:10589–10594. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chou P Y, Fasman G T. Biochemistry. 1974;13:222–228. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 48.Garnier J, Osguthorpe D J, Robson B. J Mol Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- 49.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 50.Maxwell P H, Wiesener M S, Chang G, Clifford S C, Vaux E C, Cockman M E, Wykoff C C, Pugh C W, Maher E R, Ratcliffe P J. Nature (London) 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 51.Ng R W M, Arooz T, Yam C H, Chan I W Y, Lau A W S, Poon R Y C. FEBS Lett. 1998;438:183–189. doi: 10.1016/s0014-5793(98)01299-x. [DOI] [PubMed] [Google Scholar]
- 52.Stebbins C E, Kaelin W G, Pavletich N P. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 53.Kreusch A, Pfaffinger P J, Stevens C F, Choe S. Nature (London) 1998;392:945–948. doi: 10.1038/31978. [DOI] [PubMed] [Google Scholar]
- 54.Tsuchiya H, Iseda T, Hino O. Cancer Res. 1996;56:2881–2885. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}