

Abstract
Aims: Factors responsible for the progression of non-alcoholic fatty liver disease (NAFLD) to more severe liver injury are poorly understood. In the present study, we investigated the association between immune reactions triggered by oxidative stress and stage of NAFLD.
Methods: Titres of IgG against human serum albumin adducted with malondialdehyde (MDA-HSA) or arachidonic acid hydroperoxide (AAHP) and against oxidised cardiolipin (Ox-CL) were measured in 167 NAFLD patients with steatosis only (n = 79), steatohepatitis (n = 74), or steatosis plus cirrhosis (n = 14), and in 59 age and sex matched controls.
Results: Circulating IgG against lipid peroxidation products was significantly higher (p<0.001) in NAFLD patients than in controls. Oxidative stress dependent immune responses were not associated with obesity, type 2 diabetes, or with serum cholesterol, ferritin, or aminotransferase levels. Titres of lipid peroxidation related antibodies were also independent of the extent of steatosis and were similarly distributed in patients with and without necroinflammation. In contrast, the same antibodies were significantly increased in patients with advanced fibrosis or cirrhosis. Logistic regression analysis confirmed that anti-MDA antibodies were independently associated with progression of NALFD and that NAFLD patients with titres of anti-MDA-HSA antibodies above the control threshold value had a threefold (relative risk 2.82 (95% confidence interval 1.35–5.90); p = 0.007) higher risk of having advanced fibrosis/cirrhosis than patients whose antibody titres were within the control range.
Conclusions: These results indicate that the presence of immune reactions triggered by oxidative stress can be an independent predictor of progression of NAFLD to advanced fibrosis.
Keywords: oxidative stress, immune mechanisms, liver fibrosis, steatosis
Recent epidemiological data indicate that non-alcoholic fatty liver disease (NAFLD)—ranging from simple steatosis, to non-alcoholic steatohepatitis (NASH), to cirrhosis—may be the most common liver “disease” in western countries, with a prevalence in the general population of at least 3–6%.1,2 The majority of patients with NAFLD have one or more features of the recently characterised metabolic syndrome and almost all are insulin resistant.3,4 Steatosis is therefore considered to be due to a combination of an increased hepatic supply of free fatty acids from an expanded, insulin resistant, adipose tissue mass together with the hepatic effects of the associated hyperinsulinaemia.5 What is less clear is why only 15–25% of patients with features of the metabolic syndrome progress from steatosis to NASH, characterised by necroinflammation and fibrosis,6,7 at least some of whom go on to develop cirrhosis.8 This is clearly important as elucidating the factors involved in disease progression would be a significant advance towards the rational design of treatment strategies.
Evidence derived largely from studies in various animal models has led to the notion that NASH is a disease of two “hits”, with fatty liver being the first “hit”, sensitising the liver to a variety of second “hits” capable of initiating hepatocyte injury, inflammation, and fibrosis.9 Although there are some data suggesting that gut derived endotoxins may be one of these second hits, most evidence supports a role for oxidative stress mediated lipid peroxidation in the progression of NAFLD from simple steatosis.5 Lipid peroxidation is an attractive candidate mechanism for NASH as lipid peroxidation products, including malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), potentially explain all of the histological features of NASH, including fibrosis.10,11 Studies in animal models of NAFLD have demonstrated clear evidence of increased reactive oxygen species production12–14 and lipid peroxidation.15,16 Further supporting a link between steatosis severity, lipid peroxidation, and steatohepatitis, a recent study in the MCD model has demonstrated that enhancing hepatic lipid turnover with a peroxisome proliferator activated receptor α agonist reduces oxidative stress and steatohepatitis.17
The evidence supporting a role for oxidative stress in the pathogenesis of human NASH is also increasing. Using immunohistochemical techniques, oxidation products, including 4-HNE, 8-hydroxy-deoxyguanosine, and nitrotyrosine, a marker of peroxynitrite formation, have been detected in liver biopsies from patients with NAFLD, with staining more intense in biopsies from patients with NASH compared with those with simple steatosis.4,18,19 More recently, an increase in liver and serum content of lipid peroxidation products and a decrease in plasma antioxidant ability has been documented in NAFLD patients.20,21 Indirect evidence further supporting a role for oxidative stress in “human” NASH has also come from encouraging pilot studies using various antioxidants to attenuate NASH22–24 and from a recent dietary survey reporting that obese patients with NASH consume a diet lower in antioxidants than obese patients with normal livers.25 Finally, microarray studies of liver biopsies from patients with NASH have reported that expression of mRNAs encoding several antioxidant enzymes is lower in NASH compared with normal livers or livers from patients with other liver diseases.26 Although these studies suggest that oxidative stress is associated with NAFLD in humans, evidence indicating its role in the progression of steatosis to hepatocyte injury, inflammation, and/or fibrosis is at best indirect.
Studies in our laboratories have shown that oxidative liver damage in alcohol fed rodents27 as well as in patients with alcoholic liver disease and/or chronic hepatitis C virus infection is associated with the development of circulating IgG antibodies against epitopes derived from proteins modified by lipid peroxidation products or against oxidised cardiolipin (Ox-CL) that correlate with disease severity.28–30 Moreover, a T cell mediated response towards lipid peroxidation derived antigens is also evident in patients with advanced alcoholic liver disease.31 With this rationale, we asked the following questions: (i) are immune responses mediated by oxidative stress evident in patients with non-alcoholic fatty liver and (ii) do they have any association with the presence of necroinflammation and/or severity of fibrosis?
MATERIAL AND METHODS
Patients and controls
The study included 167 consecutive patients (102 men, 65 women) with NAFLD referred from 2001 to 2003 to the Liver Unit at Newcastle Hospitals Trust (UK). All patients were negative for serological markers of hepatitis viral infection or autoimmune disease and none had any history of solvent exposure or the use of hepatotoxic drugs. Significant alcohol intake (greater than 20 g/day for men and 10 g/day for women) was excluded by structured interview administered by a specialist nurse, as described previously.32 All patients underwent full clinical and laboratory evaluation, including measurement of body mass index (BMI), waist hip ratio, and determination of insulin resistance by the homeostasis model assessment method.33 Type 2 diabetes mellitus was diagnosed according to WHO criteria.
Liver biopsy was performed in all patients and immediately fixed in formalin. Paraffin embedded sections (5 μm thick) were stained with haematoxylin/eosin, Masson’s trichrome, and periodic acid-Sciff after diastase digestion. Each biopsy was evaluated by an experienced pathologist for severity of steatosis (grade 1, <33% of hepatocytes containing fat; grade 2, 33–66%; grade 3, >66%) and the presence of necroinflammation and stage of fibrosis (stage 1, focal pericellular fibrosis confined to zone 3; stage 2, extensive pericellular/perivenular zone two thirds fibrosis with or without periportal fibrosis; stage 3, bridging fibrosis; stage 4, cirrhosis) using a modification of the scoring system devised by Brunt and colleagues.34
Fifty nine healthy controls (39 men, 20 women; mean age 47 (10) years (range 34–68)) originating from the same geographical area were recruited from hospital and university staff. All controls recorded alcohol consumption within “sensible” limits (<30 g/day for men, <20 g/day for women). All subjects gave informed consent to the analysis and the study was planned according to the guidelines of the local ethics committee. Blood samples (5 ml) were taken after an overnight fast for preparation of serum. All groups abstained from alcohol for at least 24 hours prior to blood sampling.
Antigen preparation
Human serum albumin (HSA) complexed with MDA or with reactive products of arachidonic acid oxidation (arachidonic acid hydroperoxide (AAHP)) was prepared as previously described.28,30 Briefly, 1 mg/ml HSA was reacted for two hours at 37°C with 100 mmol/l MDA in the presence of 100 mmol/l sodium cyanoborohydride or overnight at 20°C with 3 mg of arachidonic acid previously allowed to auto-oxidise for 72 hours in air. The modified HSA preparations were dialysed overnight at 4°C against phosphate buffered saline (PBS), pH 7.4. Cardiolipin (88 μg/ml in PBS) was oxidised by free radicals originating from the thermal decomposition of 1 mmol/l 2,2′′-azo-bis-(2-amidinopropane) hydrochloride (Polyscience Inc., Warrington, Pennsylvania, USA), as reported by Rigamonti and colleagues.30
Measurement of antibody titres
Polystyrene microwell plates for enzyme linked immunosorbent assay (ELISA) (Nunc-Immuno Maxi-Sorb; Nunc, S/A, Roskilde, Denmark) were coated for four hours at 37°C with 0.05 mg/ml of either modified or native HSA solubilised in 0.1 M bicarbonate buffer, pH 9.6. After incubation, solutions were removed and replaced by 0.3 ml of coating buffer containing 3% bovine serum albumin in PBS, pH 7.4. Plates were further incubated for one hour at 37°C to block non-specific binding sites. The coated wells were washed three times with PBS containing 0.25% Triton X-100. Human sera (0.20 ml, dilution 1:50 in the coating buffer) were added in duplicate and incubated for one hour at 37°C. After washing three times with PBS-0.25% Triton X-100, antibody binding was revealed using peroxidase linked goat antihuman IgG (dilution 1:6000) (Dako SPA, Milano, Italy) as previously described.28,30 Results were expressed by subtracting the background reactivity observed with unmodified HSA.
For determination of immune reactivity towards Ox-CL, ELISA plates (Nunc-Immuno Poly-Sorb) were coated by adding 30 μl of Ox-CL ethanol solution to each well and the solvent was evaporated under vacuum. The same amount of ethanol was added to reference wells. After two washes with PBS 0.3 ml, non-specific binding sites were blocked by incubation for one hour at 37°C with a 1% (v/v) solution of polyethylenglycol compound in PBS, pH 7.4. Coated wells were then washed three times with PBS. Patient and control sera (1:50 dilution in PBS) were incubated for one hour at 37°C and IgG binding was revealed, as described above. Results were expressed by subtracting the background reactivity observed in the wells treated with ethanol alone.
Competitive ELISAs were performed by preincubating overnight at 4°C human sera (1:50 dilution) with 20 μmol/l synthetic hexyl-4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde diluted in coating buffer. The same sera were also similarly incubated overnight without additions. Aliquots of the sera were then added in duplicate to ELISA plates coated with MDA-HSA and antibody binding was assayed as described above. Affinity purified rabbit polyclonal IgG directed against the 4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde (MDD) adduct were used as reference.
Materials
Malondialdehyde-bis-dimethylacetal, cardiolipin, arachidonic acid, fatty acid free human serum albumin (fraction V), polyethylenglycol compound, and sodium cyanoborohydride were supplied by Sigma Chemical Co. (St Louis, Missouri, USA). All other chemicals were of analytical grade and were supplied by Merck (Darmstad, Germany). Hexyl-4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde and affinity purified rabbit polyclonal IgG against the same antigen were a kind gift from Dr G Thiele (University of Nebraska, Omaha, USA)
Data analysis and statistical calculations
Statistical analyses were performed by SPSS statistical software (SPSS Inc., Chicago, Illinois, USA) using the one way ANOVA test or the Kruskal-Wallis test. Confidence intervals (CI) were calculated using CIA software (by T Bryant, University of Southampton, UK). Relative risk and Fisher’s exact tests were used for comparison of frequency data. Significance was taken at the 5% level. The independent effect of significant variables was assessed using stepwise logistic regression analysis. Cut off values were calculated as 97.5th percentiles in the control population after log transformation of the values and assessment of the distribution normality by the Shapiro-Francia test.
RESULTS
Patient characteristics
Of 167 patients included in the study, 79 had steatosis only, 74 had steatosis, necroinflammation, and various degree of fibrosis (NASH), and 14 had steatosis plus cirrhosis. The biochemical and clinical features of the three groups are shown in table 1 ▶. Consistent with results from previous studies,7,8,35,36 patients with cirrhosis were older than those with steatosis or NASH, and patients with NASH and cirrhosis had a higher BMI and were more likely to be diabetic than those with steatosis only. Patients with NASH had higher serum transaminases than patients with steatosis. Serum bilirubin, alkaline phosphatase, γ-glutamyl transpeptidase levels, and aspartate aminotransferase/alanine aminotransferase (AST/ALT) ratio were higher and serum albumin levels lower in patients with cirrhosis compared with the two other groups of patients.
Table 1.
Clinical and biochemical characterisation of patients with non-alcoholic fatty liver disease
| Steatosis | NASH | Steatosis and cirrhosis | |
| No of patients (M/F) | 79 (54/25) | 74 (39/35) | 14 (8/6) |
| Age (y) | 51.5 (13.7)** | 53.6 (13.3)** | 60.3 (9.9) |
| Body mass index (kg/m2) | 32.4 (5.9)*† | 34.5 (4.7) | 36.7 (4.4) |
| Diabetes (%) | 12 (15) | 28 (38) | 9 (64) |
| AST (U/l) | 33.8 (15.1)**†† | 47.8 (23.6) | 50.3 (28.0) |
| ALT (U/L) | 56.3 (34.2)† | 73.8 (50.6) | 53.5 (51.9) |
| AST/ALT ratio | 0.70 (0.29)*** | 0.73 (0.24)*** | 1.28 (0.64) |
| γ-glutamyl transpeptidase (U/l) | 85.6 (54.7)** | 73.3 (49.0)*** | 170.9 (239.3) |
| Alkaline phosphatase (U/l) | 86.0 (29.6)** | 86.6 (33.0)** | 107 (41.4) |
| IgG | 10.91 (2.27)** | 10.99 (3.29)** | 15.51 (4.32) |
| Bilirubin (μmol/l) | 11.9 (6.3)* | 10.7 (6.6)* | 16.0 (9.6) |
| Ferritin (μg/l) | 153.2 (112.3) | 175.9 (106.2) | 175.0 (193.8) |
| Cholesterol (mmol/l) | 6.0 (1.5)***† | 5.6 (1.1)† | 4.4 (1.1) |
| Triglycerides (mmol/l) | 2.8 (1.8) | 3.0 (2.0) | 2.1 (0.9) |
| Albumin (g/l) | 45.5 (2.8)*** | 44.7 (3.5)*** | 39.7 (5.6) |
Statistically significant versus steatosis plus cirrhosis: *p<0.05; **p<0.01; ***p<0.0001. Statistically significant versus NASH: †p<0.05; ††p<0.0001.
AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Lipid peroxidation related antibodies in NAFLD patients and controls
Figure 1 ▶ shows that the titres of IgG against MDA-HSA or AAHP-HSA and against Ox-CL were significantly higher (p<0.0001) in NAFLD patients than in controls. Furthermore, approximately a third (29–39%) of NAFLD patients had MDA-HSA, AAHP-HSA, and OxCL IgG titres above the 97.5th percentile in controls—the “threshold” defining a “positive” titre (fig 1 ▶). These differences could not be accounted for by differences in circulating IgG, which was comparable in NAFLD and control subjects (11.36 (3.21) v 10.63 (3.15); 95% CI −1.68 to 0.22). Moreover, there was a poor correlation between individual reactivity against the different lipid peroxidation derived antigens and serum IgG content (r ranged from 0.19 and 0.35). Titres of the three antibodies did not correlate with BMI, serum cholesterol, triglyceride or ferritin, any individual liver blood test (alkaline phosphatase, alanine/aspartate transaminase, γ-glutamyl transpeptidase), or the AST/ALT ratio. In addition, titres of lipid peroxidation derived antibodies were not different between NAFLD patients with (n = 49) and without (n = 118) type 2 diabetes (od490 nm for anti-MDA-HSA IgG 0.36 (0.12) v 0.35 (0.15), 95% CI −0.053 to 0.034; od490 nm for anti-AAPH-HSA IgG 0.08 (0.06) v 0.10 (0.07), 95% CI −0.001 to 0.041; od490 nm for anti-OxCL IgG 0.13 (0.10) v 0.13 (0.11), 95% CI −0.035 to 0.035).
Figure 1.

Titres of IgG against human serum albumin adducted with malondialdehyde (MDA-HSA) and arachidonic acid hydroperoxides (AAHP-HSA) or against oxidised cardiolipin (Ox-CL) in patients with non-alcoholic fatty liver disease (NAFLD) and in healthy controls. Sera were tested at a 1:50 dilution in microplate ELISA plates coated with different antigens and revealed with peroxidase linked goat antihuman IgG antiserum. Results are expressed as optical density (od) at 490 nm after subtracting the background reactivity of each serum. Boxes show values within 25th and 75th percentiles, horizontal bar represents the median, 80% of values are between the extremities of the vertical bars (10th–90th percentiles), and extreme values are represented by individual symbols. Values under the boxes represent the percentage of subjects with IgG titres above the cut off value, calculated on the 97.5th percentile of the control population. Statistical significance, NAFLD versus controls: MDA-HSA p<0.0001 (95% confidence interval (CI) −0.169 to −0.11); AAHP-HSA, p<0.0001 (95% CI −0.042 to −0.018); Ox-CL p<0.0001 (95% CI −0.059 to −0.020).
Oxidative stress mediated immune response and histology in patients with NAFLD
When lipid peroxidation induced immune responses were investigated in relation to liver histology, no significant difference in antibody titres was observed in relation to the severity of steatosis (fig 2 ▶). Similarly, the titres of anti-MDA-HSA, anti-AAHP-HSA, and anti-Ox-CL IgG were comparable in patients with steatosis only and those with steatohepatitis (fig 2 ▶). Patients with and without necroinflammation were also not different in terms of frequency of “positive” lipid peroxidation related antibody titres (fig 2 ▶).
Figure 2.
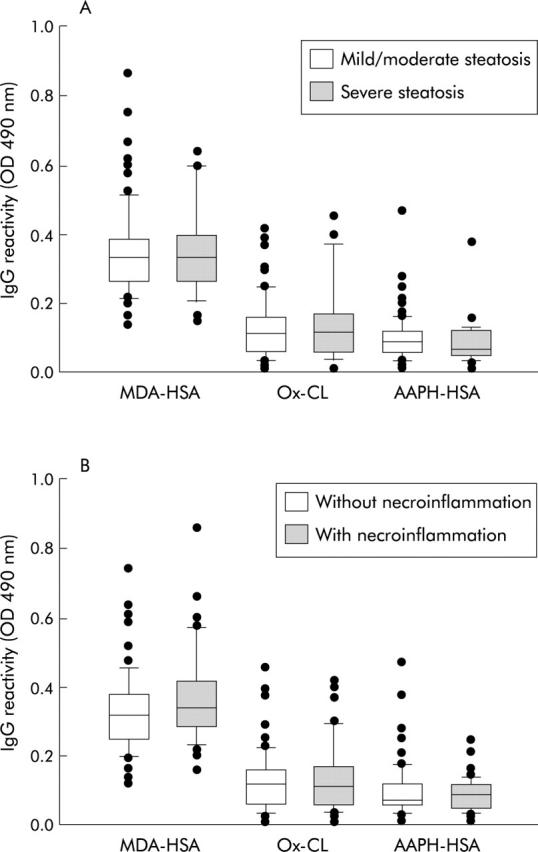
Distribution of IgG against human serum albumin adducted with malondialdehyde (MDA-HSA) and arachidonic acid hydroperoxides (AAHP-HSA) or against oxidised cardiolipin (Ox-CL) in patients with non-alcoholic fatty liver disease (NAFLD) according to the extent of steatosis (A) or the presence of necroinflammation (B). Steatosis was scored as mild/moderate (grades 1–2) or severe (grade 3), as reported in the methods section. Sera were tested at 1:50 dilution in microplate enzyme linked immunoabsorbent assay plates and antibody binding was revealed with peroxidase linked goat antihuman IgG antiserum. Results are expressed as optical density (od) at 490 nm after subtracting the background reactivity of each serum. Boxes show values within 25th and 75th percentiles, horizontal bar represents the median, 80% of values are between the extremities of the vertical bars (10th–90th percentiles), and extreme values are represented by individual symbols.
To investigate the possible relationship between oxidative stress mediated immune response and the degree of fibrosis, NAFLD patients were regrouped according to stage of fibrosis. Eighty four subjects (50%) had no evidence of fibrosis, 35 (21%) had stage 1 fibrosis, 19 (11%) stage 2, 15 (9%) stage 3, and 14 (8%) stage 4 (cirrhosis). As shown in fig 3 ▶, patients without fibrosis (score 0) or with mild fibrosis (scores 1 and 2) had titres of anti-MDA-HSA and anti-Ox-CL antibodies significantly lower than those with advanced fibrosis (scores 3 and 4). In this latter group the percentage of subjects positive for two or more lipid peroxidation related antibodies (46%) was also significantly (95% CI 1–38%; p<0.05) higher than in subjects without fibrosis or with mild fibrosis only (27%). NAFLD patients with anti-MDA-HSA antibodies above the control threshold value had a threefold higher risk of having advanced fibrosis or cirrhosis (relative risk 2.82; 95% CI 1.35–5.90; p = 0.007) compared with patients whose antibody titres were within the control range.
Figure 3.
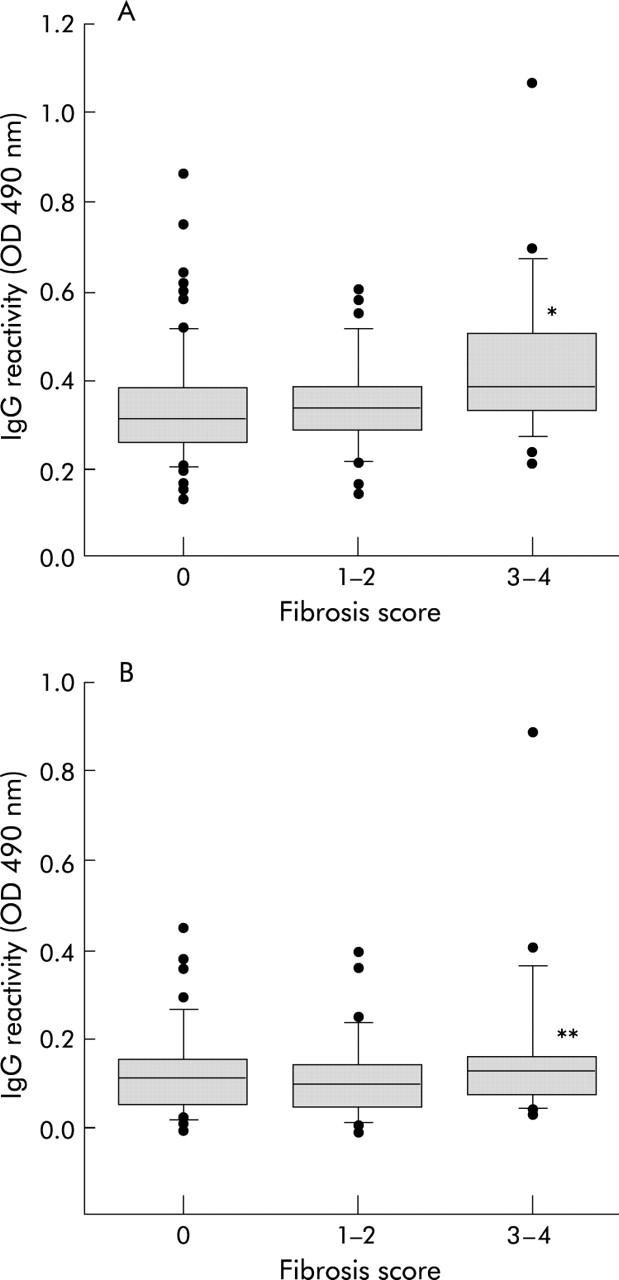
Titres of IgG against human serum albumin adducted with malondialdehyde (MDA-HSA) (A) or against oxidised cardiolipin (Ox-CL) (B) in patients with non-alcoholic fatty liver disease according to fibrosis stage. Sera were tested at a 1:50 dilution in microplate enzyme linked immunoabsorbent assay plates coated with different antigens and revealed with peroxidase linked goat antihuman IgG antiserum. Boxes show values within 25th and 75th percentiles, horizontal bar represents the median, 80% of values are between the extremities of the vertical bars (10th–90th percentiles), and extreme values are represented by individual symbols. Statistical significance: *p<0.05 versus no fibrosis (95% confidence interval (CI) 0.01 to 0.15) and fibrosis stages 1–2 (95% CI 0.01 to 0.16); **p<0.05 versus no fibrosis (95% CI 0.01 to 0.12) and fibrosis stages 1–2 (95% CI 0.01–0.11).
Previous studies have established obesity (BMI), diabetes, age, and AST/ALT ratio >1 as independent predictors of disease progression in NASH patients.7,35,36 To investigate whether the magnitude of oxidative stress might represent an additional predictor of the presence of advanced fibrosis, the above variables were investigated in combination with the presence of anti-MDA antibodies. Univariate analysis confirmed that the presence of advanced fibrosis (stages 3, 4) in our NAFLD patients was also associated with age >45 years and with the presence of diabetes or an AST/ALT ratio >1 (p value ranging from 0.015 to <0.0001). However, following stepwise logistic regression, only diabetes, AST/ALT ratio >1, and anti-MDA antibodies were found to be independent predictors of severe fibrosis in NAFLD (table 2 ▶) and their combination was able to explain 39% of the variability in disease severity.
Table 2.
Logistic regression analysis of the association between independent predictors and the presence of severe fibrosis in patients with non-alcoholic fatty liver disease
| Variable | Regression coefficient (B) | SE of regression coefficient | Wald value | E ex (B) | p Value (95% CI) |
| Diabetes | 2.004 | 0.583 | 11.824 | 7.74 | 0.001 (2.37–23.26) |
| AST/ALT ratio >1 | 1.802 | 0.595 | 9.183 | 6.06 | 0.002 (1.89–19.46) |
| Anti-MDA antibodies | 4.990 | 1.831 | 7.430 | 146.93 | 0.006 (4.06–5312.9) |
| Constant | −4.843 | ||||
| Nagelkerke’s r2 | 0.390 |
Overall fit of the model χ2 33.832 with 3 DF; p<0.0005.
Percent of correct predictions 87.3%
AST/ALT, aspartate aminotransferase/alanine aminotransferase; MDA, malondialdehyde.
Characterisation of the reactivity against MDA adducts associated with progression of NAFLD
In view of the association between the IgG response against MDA adducts and progression of NAFLD to fibrosis, we have attempted to characterise the antigen specificity of the antibodies involved. Previous studies have demonstrated that MDD (fig 4 ▶), originating from the reaction of MDA with the epsilon amino group of lysine, is one of the main MDA-protein adducts and has strong antigenic properties.37 Competition experiments using 28 NAFLD sera with high reactivity towards MDA-HSA and 13 control sera showed that preincubation with 20 μmol/l of synthetic hexyl-MDD derivative abolished the reactivity of affinity purified rabbit IgG against the MDD adduct and reduced by approximately 40% the recognition of MDA-HSA by NAFLD sera (od490 nm 0.389 (0.153) v 0.178 (0.11); 95% CI −0.29 to −0.14; p<0.0001) without significantly affecting that of control sera (od490 nm 0.256 (0.196) v 0.194 (0.17); 95% CI −0.21 to −0.08) (fig 4 ▶). Moreover, following preadsorption with hexyl-MDD, no significant difference in reactivity towards MDA-HSA was appreciable between NAFLD and control sera (od490 nm 0.178 (0.11) v 0.194 (0.17); 95% CI −0.091 to 0.124), suggesting that antibodies directed towards MDA adducts specifically recognise dihydropyridine epitopes.
Figure 4.

Characterisation of the antibody response towards malondialdehyde (MDA) adducts in patients with non-alcoholic fatty liver disease (NAFLD). Sera of 28 NAFLD patients with reactivity against MDA adducts and 13 sera from healthy controls (1:50 dilution) were preincubated overnight at 4°C with 20 μmol/l synthetic hexyl-4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde (MDD) diluted in coating buffer. The same sera were also similarly incubated overnight without additions. Aliquots of the sera were then added in duplicated to enzyme linked immunoabsorbent assay plates coated with MDA-human serum albumin (HSA) and the antibody binding was assayed as described in the methods section. Affinity purified rabbit polyclonal IgG directed against the MDD adduct was used as reference. The insert show the chemical structure of the hexyl-MDD adduct.
DISCUSSION
The mechanisms responsible for progression of NAFLD to more severe liver injury are still poorly understood. The “two hit” model proposes that fat accumulation—the “first hit”—sensitises the liver to the injurious effects of one or more additional factor(s)—“second hits”—that lead to the development of steatohepatitis and fibrosis.9 In this model, oxidative stress, arising as a result of increased formation of reactive oxygen species during fatty acid oxidation by mitochondria and peroxisomes and by the induction of cytochrome P450 isoenzymes (CYP2E1 and CYP4A), coupled with the mitochondrial effects of tumour necrosis factor α, has been proposed as one such factor.4,5,12,17 To date, however, evidence supporting this hypothesis has been derived largely from animal models. In the present study, we measured titres of circulating antibodies against lipid peroxidation products and demonstrated that approximately a third of patients with NAFLD had titres of these antibodies above the 97.5th percentile of healthy controls. Furthermore, NAFLD patients with anti-MDA antibodies above the control threshold value had an approximately threefold increased risk of developing advanced fibrosis or cirrhosis compared with subjects with antibody titres within the control range. These data provide strong evidence that oxidative stress promotes immune responses in a proportion of patients with NAFLD and that these responses are associated with the presence of advanced disease.
Partial characterisation of the epitope specificity of anti-MDA antibodies associated with NAFLD shows that methyl-1,4-dihydropyridine-3,5-dicarbaldehyde adducts play an important role. It is noteworthy that the same di-hydropyridine epitopes are responsible for a specific humoral immune response towards adducts generated by the condensation of MDA and acetaldehyde, also known as malondialdehyde-acetaldehyde adducts,38 observed in both alcohol fed animals39 and patients with alcoholic liver disease.40 At present, however, the mechanisms leading to the generation of di-hydropyridine adducts in NAFLD are not known.
The lack of any correlation between the detection of lipid peroxidation related antibodies and the presence of necroinflammation in human NAFLD is consistent with other reports that have detected lipid peroxidation4,18,20 in liver biopsies of NAFLD patients with steatosis only. This suggests that oxidative stress is unlikely to be either a necessary cause or an effect of necroinflammation in NAFLD. It is noteworthy that the frequency (30–40%) of patients with antibodies towards lipid peroxidation antigens observed in this study is much lower than the increase in liver MDA and 4-HNE content (more that 90%) recently reported by Loguercio and colleagues,20 suggesting that dietary or genetic factors25,41 are required for the onset of the oxidative stress dependent immune response in NAFLD patients.
The concept of oxidative stress as an important mechanism of hepatic fibrosis is now well established.42 A more specific profibrogenic role for lipid peroxidation protein adducts has been suggested by recent studies demonstrating that human hepatic stellate cells (HSC) have the capacity to specifically recognise MDA modified proteins through an interaction with CD36 scavenger receptors.43 Such an interaction not only stimulates the synthesis of fibronectin and collagen by HSC, but may also be important in promoting an immune response to the adducts, as human HSC have recently been shown to act as antigen presenting cells capable of stimulating lymphocyte proliferation.44 This dual effect of lipid peroxidation protein adducts on HSC offers a potential explanation for the association between anti-MDA IgG titres and fibrosis observed in the present study. A role for immune response triggered by oxidative stress in the progression of NAFLD is at present speculative although it is worth noting that an accumulating body of evidence supports its role in the pathogenesis of alcoholic liver disease31,45 as well as in worsening of chronic hepatitis C by alcohol intake.30 Moreover, further support of a role of the immune response in liver fibrosis has been provided by a recent report that lymphocyte destruction by subletal irradiation reduces liver fibrosis in mice treated with CCl4 or thioacetamide, while the transfer of CD8+ lymphocytes from CCl4 treated mice to immunodeficient SCID mice leads to fibrogenic activation of HSC.46
In the recent years there has been a growing interest in establishing reliable predictors for the presence of advanced NAFLD in patients presenting with classical risk factors, abnormal liver biochemistry, and ultrasound evidence of steatosis.7,35,36 Given our observation that anti-MDA titres were significantly higher in patients with advanced fibrosis, we examined their predictive value for clinically significant stage 3/4 fibrosis in a regression model, including the previously described predictive clinical and laboratory factors. This analysis demonstrated that diabetes, AST/ALT ratio >1, and anti-MDA antibodies were independently associated with severe fibrosis explaining, in combination, 39% of the variability in fibrosis score, suggesting that anti-MDA titres may be a clinically useful easy to measure serum marker of advanced NAFLD. The observation that anti-MDA titres and diabetes were independent predictors of fibrosis is important given previous reports of antibodies against oxidised lipoproteins in the sera of patients with type 2 diabetes.47,48 This excluded the possibility that the association between anti-MDA and advanced fibrosis is explained by the confounding effect of diabetes.
In conclusion, these results demonstrate that lipid peroxidation related antibodies are present in a proportion of patients with NAFLD and are associated with advanced fibrosis, but not with steatosis severity or necroinflammation. This association provides further support for the suggestions that oxidative stress is involved in the progression of NAFLD to fibrosis and is a rational target for therapy. If confirmed in prospective studies, evaluation of IgG reactivity towards dihydropyridine MDA adducts may become a useful non-invasive serum marker both for the presence of advanced disease and, more importantly, for identification of NAFLD patients at risk of progression, potentially with the most to gain from targeted antioxidant therapy.22–24
Acknowledgments
This work was supported by a grants from the Italian Ministry for University and Scientific and Technological Research (Research Program: 2002062883 Malattie epatiche steatosiche non alcoliche; fattori e meccanismi di danno epatico e progressione) and the Regione Piemonte.
Abbreviations
AAHP-HSA, arachidonic acid hydroperoxide adduct with human serum albumin
ELISA, enzyme linked immunoabsorbent assay
4-HNE, 4-hydroxynonenal
HSA, human serum albumin
HSC, human hepatic stellate cells
MDD, 4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde
NAFLD, non-alcoholic fatty liver disease
NASH, non-alcoholic steatohepatitis
MDA-HSA, malondialdehyde adduct with human serum albumin
Ox-CL, oxidised cardiolipin
BMI, body mass index
PBS, phosphate buffered saline
AST/ALT, aspartate aminotransferase/alanine aminotransferase
Conflict of interest: None declared.
REFERENCES
- 1.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol 2003;98:960–7. [DOI] [PubMed] [Google Scholar]
- 2.Ruhl CE, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterol 2003;124:71–9. [DOI] [PubMed] [Google Scholar]
- 3.Marchesini G, Bugianesi E, Forlani G, et al. Non alcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatol 2003;37:917–23. [DOI] [PubMed] [Google Scholar]
- 4.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Non-alcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterol 2001;120:1183–92. [DOI] [PubMed] [Google Scholar]
- 5.Day CP. Pathogenesis of steatohepatitis. Best Practice Res Clin Gastroenterol 2002;16:663–78. [DOI] [PubMed] [Google Scholar]
- 6.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatol 1990;12:1106–10. [DOI] [PubMed] [Google Scholar]
- 7.Dixon JB, Bhathal PS, O’Brian PE. Non-alcoholic fatty liver disease: predictors of non-alcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterol 2001;121:91–100. [DOI] [PubMed] [Google Scholar]
- 8.Caldwell SH, Oelsner DH, Iezzoni JC, et al. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatol 1999;29:664–70. [DOI] [PubMed] [Google Scholar]
- 9.Day CP, James OFW. Steatohepatitis: a tale of two hits? Gastroenterology 1998;114:842–5. [DOI] [PubMed] [Google Scholar]
- 10.Pessayre D, Berson A, Fromenty B, et al. Mitochondria in steatohepatitis. Semin Liver Dis 2001;21:57–69. [DOI] [PubMed] [Google Scholar]
- 11.Zamara E, Novo E, Marra F, et al. 4-hydroxynonenenal as a selective pro-fibrogeneic stimulus for activated human hepatic stellate cells. J Hepatol 2004;40:60–8. [DOI] [PubMed] [Google Scholar]
- 12.Yang SQ, Zhu H, Li Y, et al. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys 2000;378:259–68. [DOI] [PubMed] [Google Scholar]
- 13.Hensley K, Kotake Y, Sang H, et al. Dietary choline restriction causes complex I dysfunction and increased H2O2 generation in liver mitochondria. Carcinogen 2000;21:983–9. [DOI] [PubMed] [Google Scholar]
- 14.Laurent A, Nicco C, Tran Van Nhieu J, et al. Pivotal role of superoxide anion and benefical effect of antioxidant molecules in murine steatohepatitis. Hepatol 2004;39:1277–85. [DOI] [PubMed] [Google Scholar]
- 15.Lequercq IA, Farrell GC, Field J, et al. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine non-alcoholic steatohepatitis. J Clin Invest 2000;105:1067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Letteron P, Fromenty B, Terris B, et al. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J Hepatol 1996;24:200–8. [DOI] [PubMed] [Google Scholar]
- 17.Ip E, Farrell GC, Robertson G, et al. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003;38:123–32. [DOI] [PubMed] [Google Scholar]
- 18.Seki S, Kitada T, Yamada T, et al. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver disease. J Hepatol 2002;37:56–62. [DOI] [PubMed] [Google Scholar]
- 19.Chalasani N, Deeg MA, Crabb DW. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with non-alcoholic steatohepatitis. Am J Gastroenterol 2004;99:1497–1502. [DOI] [PubMed] [Google Scholar]
- 20.Loguercio C, De Simone T, D’Auria MV, et al. Non-alcoholic fatty liver disease: a multicentre clinical study by the Italian Association for the Study of the Liver. Dig Liver Dis 2004;36:398–405. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Monzon C, Martin-Perez E, Iacono OL, et al. Characterization of pathogenic and prognostic factors of non-alcoholic steatohepatitis associated with obesity. J Hepatol 2000;33:716–24. [DOI] [PubMed] [Google Scholar]
- 22.Merat S, Malekzadeh R, Sohrabi MR, et al. Probucol in the treatment of non-alcoholic steatohepatitis: a double-blind randomized controlled study. J Hepatol 2003;38:414–18. [DOI] [PubMed] [Google Scholar]
- 23.Lavine JE. Vitamin E treatment of non-alcoholic steatohepatitis in children: A pilot study. J Pediatr 2000;136:734–8. [PubMed] [Google Scholar]
- 24.Harrison SA, Torgerson S, Hayashi P, et al. Vitamin E and vitamin C treatment improves fibrosis in patients with non-alcoholic steatohepatitis. Am J Gastroenterol 2003;98:2485–90. [DOI] [PubMed] [Google Scholar]
- 25.Musso G, Gambino R, De Michieli F, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003;37:909–16. [DOI] [PubMed] [Google Scholar]
- 26.Sreekumar R, Rosado B, Rasmussen D, et al. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology 2003;38:244–51. [DOI] [PubMed] [Google Scholar]
- 27.Albano E, Clot P, Morimoto M, et al. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology 1996;23:155–63. [DOI] [PubMed] [Google Scholar]
- 28.Rolla R, Vay D, Mottaran E, et al. Anti-phospholipid antibodies associated with alcoholic liver disease specifically recognize oxidized phospholipids. Gut 2001;49:852–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mottaran E, Stewart SF, Rolla R, et al. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic Biol Med 2002;32:38–48. [DOI] [PubMed] [Google Scholar]
- 30.Rigamonti C, Mottaran E, Reale E, et al. Moderate alcohol consumption increases oxidative stress in patients with chronic hepatitis C. Hepatology 2003;38:42–9. [DOI] [PubMed] [Google Scholar]
- 31.Stewart SF, Vidali M, Day CP, et al. Oxidative stress as a trigger for cellular immune response in patients with alcoholic liver disease. Hepatology 2004;39:197–203. [DOI] [PubMed] [Google Scholar]
- 32.Grove J, Daly AK, Brown AStJM, et al. The Rsa I polymorphism of CYP2E1 and susceptibility to alcoholic liver disease in Caucasians: effect on age of presentation and dependence on alcohol dehydrogenase genotype. Pharmacogenetics 1998;8:335–42. [DOI] [PubMed] [Google Scholar]
- 33.Haffner SM, Kennedy E, Gonzalez C, et al. A prospective analysis of the HOMA model. The Mexico City Diabetes Study. Diabet Care 1996;19:1138–41. [DOI] [PubMed] [Google Scholar]
- 34.Brunt EM, Jannery CJ, Di Bisceglie AM, et al. Non-alcoholic steatohepatitis: a proposal for grading and staging the histologic lesions. Am J Gastroenterol 1999;94:2467–76. [DOI] [PubMed] [Google Scholar]
- 35.Angulo P, Keach JC, Batts KP, et al. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999;30:1356–62. [DOI] [PubMed] [Google Scholar]
- 36.Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology 2000;118:1117–23. [DOI] [PubMed] [Google Scholar]
- 37.Yamada S, Kumazawa S, Ishii T, et al. Immunological detection of lipofuscin-like fluorophore derived from malondialdehyde and lysine. J Lipid Res 2001;42:1187–96. [PubMed] [Google Scholar]
- 38.Tuma DJ, Thiele GM, Xu D, et al. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology 1996;23:872–80. [DOI] [PubMed] [Google Scholar]
- 39.Xu D, Thiele GM, Beckenhauer JL, et al. Detection of circulating antibodies to malondialdehyde-acetaldehyde adducts in ethanol-fed rats. Gastroenterol 1998;115:686–92. [DOI] [PubMed] [Google Scholar]
- 40.Rolla R, Vay D, Mottaran E, et al. Detection of circulating antibodies against malondialdehyde-acetaldehyde adducts in patients with alcoholic liver disease. Hepatology 2000;31:878–84. [DOI] [PubMed] [Google Scholar]
- 41.Chikako N, Zhang SP, Raynor VJ, et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J Hepatol 2004;40:781–6. [DOI] [PubMed] [Google Scholar]
- 42.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol 2001;35:297–306. [DOI] [PubMed] [Google Scholar]
- 43.Schneiderhan W, Schmid-Kotsas A, Zhao J, et al. Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology 2001;34:729–37. [DOI] [PubMed] [Google Scholar]
- 44.Viñas O, Bataller R, Sancho-Bru P, et al. Human hepatic stellate cells show features of antigen presenting cells and stimulate lymphocyte proliferation. Hepatology 2003;38:919–29. [DOI] [PubMed] [Google Scholar]
- 45.Albano E. Free radical mechanisms in immune reactions associated with alcoholic liver disease. Free Radic Biol Med 2002;32:110–14. [DOI] [PubMed] [Google Scholar]
- 46.Safadi R, Ohta M, Alvarez CE, et al. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004;127:870–82. [DOI] [PubMed] [Google Scholar]
- 47.Bellomo G, Maggi E, Poli M, et al. Autoantibodies against oxidatively modified low density in NIDDM. Diabetes 1995;44:60–6. [DOI] [PubMed] [Google Scholar]
- 48.Mezzetti A, Cipollone F, Cuccurullo F. Oxidative stress and cardiovascular complications in diabetes: isoprostanes as new markers on an old paradigm. Cardiovasc Res 2000;47:475–88 (rrratum in Cardiovasc Res 2003;57: 869. [DOI] [PubMed] [Google Scholar]