

Summary
A balance in the activities of the Ipl1 Aurora kinase and the Glc7 phosphatase is essential for normal chromosome segregation in yeast. We report here that this balance is modulated by the Set1 methyltransferase. Deletion of SET1 suppresses chromosome loss in ipl1-2 cells. Conversely, combination of SET1 and GLC7 mutations is lethal. Strikingly, these effects are independent of previously defined functions for Set1 in transcription initiation and histone H3 methylation. We find that Set1 is required for methylation of conserved lysines in a kinetochore protein, Dam1. Biochemical and genetic experiments indicate that Dam1 methylation inhibits Ipl1-mediated phosphorylation of flanking serines. Our studies demonstrate that Set1 has important, unexpected functions in mitosis. Moreover, our findings suggest that antagonism between lysine methylation and serine phosphorylation is a fundamental mechanism for controlling protein function.
Introduction
Proper chromosome segregation requires that centromeres of sister chromatids be attached to microtubules emanating from opposite spindle poles. These attachments are mediated by kinetochores, which are assembled from at least 60 different proteins in S. cerevisiae (Cheeseman et al., 2002a, 2002b; Westermann et al., 2005). Interactions among these proteins and between kinetochores and microtubules are highly dynamic, in part to allow resolution of improper monopolar sister chromatid attachments and reformation of proper bipolar attachments (Ducat and Zheng, 2004). These interactions are largely controlled by cycles of phosphorylation and dephosphorylation of specific kinetochore proteins (Cheeseman et al., 2002a, 2002b; Courtwright and He, 2002).
Aurora kinases play a key role in the organization of the kinetochore and are essential for normal chromosome segregation in all organisms from yeast to humans (Ducat and Zheng, 2004). Chromosome loss and aberrant centrosome replication occur in cells that exhibit abnormal Aurora kinase expression. Overexpression of Aurora A or Aurora B is often observed in colorectal cancers and in several cancer cell lines (Meraldi et al., 2004), highlighting the importance of Aurora kinase regulation to genome stability and tumor suppression.
Ipl1 is the only member of this highly conserved kinase family in S. cerevisiae. Deletion of IPL1 is lethal and ipl1 point mutations lead to abnormal chromosome segregation and aneuploidy (Chan and Botstein, 1993; Francisco and Chan, 1994). Phosphorylation of kinetochore proteins by Ipl1 modulates protein-protein interactions, thereby governing kinetochore integrity (Cheeseman et al., 2002a, 2002b; Courtwright and He, 2002). Ipl1-mediated phosphorylation events are required both for turnover of improper monopolar attachments of sister kinetochores as well as the tension-sensing component of the mitotic checkpoint (Ducat and Zheng, 2004). Ipl1 also functions in spindle disassembly (Buvelot et al., 2003).
Dam1 is an important mitotic substrate for Ipl1 (Cheeseman et al., 2002a; Courtwright and He, 2002). A complex containing Dam1 and eight other proteins oligomerizes into rings around microtubules (Westermann et al., 2005) and is important both for proper chromosome segregation as well as maintaining the integrity of the spindle (Courtwright and He, 2002). Ala-nine substitution of multiple serines in Dam1 that are phosphorylated by Ipl1 yields an abnormal chromosome segregation phenotype similar to that caused by ipl1 mutations (Cheeseman et al., 2002a). Dephosphorylation of Dam1 and other Ipl1 substrates by the Glc7 phosphatase is also essential for yeast viability (Francisco et al., 1994; Sassoon et al., 1999).
Ipl1 and Glc7 regulate the phosphorylation of histone H3 at serine 10 (S10) (Hsu et al., 2000). H3 S10 mutations have no mitotic consequence in S. cerevisiae, but S10 phosphorylation serves as a mitotic marker in many organisms (Crosio et al., 2002; Wei et al., 1999). Histones are subject to multiple other posttranslational modifications, including acetylation, methylation, and ubiquitylation (Zhang and Reinberg, 2001). Histone-modification patterns may constitute an informational code that governs both the structure of chromatin and interactions of nonhistone regulatory proteins with nucleosomes (Jenuwein and Allis, 2001). Interestingly, phosphorylation of S10 can enhance acetylation of K14 in H3 (Cheung et al., 2000; Lo et al., 2000), abolish acetylation of K9 (Edmondson et al., 2002), and inhibit methylation of K9 (Rea et al., 2000). Methylation of H3 K9, in turn, inhibits phosphorylation of S10 (Rea et al., 2000). Such interplay illustrates the potential for regulatory crosstalk between different posttranslational modifications within an individual protein (Zhang and Reinberg, 2001).
H3 methylation at K4 is mediated by the Set1 protein in S. cerevisiae (Briggs et al., 2001), which affects both gene activation and repression (Briggs et al., 2001; Krogan et al., 2002; Noma and Grewal, 2002). Set1 is part of a multisubunit assembly termed COMPASS that interacts with the C-terminal domain of the large subunit of RNA polymerase II through association with the Paf1 complex (Tenney and Shilatifard [2005], for review). In addition, H3 K4 dimethylation and COMPASS recruitment to the 5′ coding regions of genes requires ubiquitylation of histone H2B at K123 by Rad6-Bre1, which is also dependent on Paf1 (Tenney and Shilatifard, 2005).
We report here that deletion of SET1 suppresses the abnormal chromosome segregation normally observed in cells bearing a temperature sensitive ipl1 mutation. Surprisingly, this suppression is not linked to methylation of H3 and is mimicked only partially by mutation of H3 K4. Suppression of ipl1 is not observed upon loss of Paf1 or mutation of H2B K123, indicating that Set1 has unanticipated and important functions during mitosis that are independent of its roles in transcription initiation and early elongation. Our experiments indicate that Set1 methylates Dam1 and that a proper balance of Dam1 methylation and phosphorylation is critical for normal chromosome segregation and cell viability.
Results
Mutation of SET1 Suppresses the Temperature-Sensitive Phenotype of ipl1-2
To determine if the functions of the Set1 methyltransferase and the Ipl1 kinase are linked in vivo, we combined a deletion of SET1 with ipl1-2, a well-characterized temperature-sensitive allele of IPL1 (Chan and Botstein, 1993; Francisco and Chan, 1994). ipl1-2 cells are viable at 25°C but exhibit defective growth from 30°C to 32.5°C and are not viable at higher temperatures (Figure 1).
Figure 1.

- Yeast strains with the indicated genotypes were serially diluted 10-fold, spotted onto YPD medium, and grown at indicated temperatures for 2 days. Growth of four independent ipl1-2 set1 Δ colonies is shown.
- Plate spot assays performed as above to test the ability of the set1 G951S mutation to suppress the temperature sensitivity of ipl1-2.
- The stability of an artificial minichromosome marked by the SUP11 tRNA gene, which suppresses a defective ade2 allele, was measured in the indicated wild-type and mutant strains. Colony color was scored after 4 days of growth at 25°C. Shown are the averages of two independent experiments. Error bars indicate standard deviations.
Deletion of SET1 alone does not affect cell growth at 32.5°C or 34°C (Figure 1A). Remarkably, deletion of SET1 greatly improves growth of ipl1-2 cells at 30°C and 32.5°C (Figure 1A). Analysis of 10-fold serial dilutions in a plate spot assay indicates that set1 Δ ipl1-2 cells grow at least 100 times better than ipl1-2 single mutants at these elevated temperatures. Importantly, the double mutant cells do not survive at 34°C or higher temperatures, indicating that loss of Set1 suppresses some defective Ipl1 functions but does not bypass the need for Ipl1.
The suppression of ipl1-2 observed in set1 Δ cells might reflect the loss of Set1 catalytic activity or compromised integrity of the COMPASS complex (Krogan et al., 2002) in the absence of Set1. To distinguish these possibilities, we mutated G951 in Set1 to S. The G951S mutation severely reduces Set1 catalytic activity without influencing COMPASS integrity (Nagy et al., 2002). We found that this mutation suppresses the temperature sensitivity of ipl1-2 at 30°C and 32.5°C (Figure 1B). Therefore, loss of Set1 methyltransferase activity is sufficient for suppression of ipl1-2.
Chromosome Loss in ipl1-2 Cells Is Suppressed by SET1 Deletion
To determine whether loss of SET1 suppresses the high frequency of chromosome loss observed in ipl1-2 cells (Chan and Botstein, 1993; Francisco and Chan, 1994), we compared the stability of a nonessential marker chromosome carrying the SUP11 suppressor tRNA in isogenic wild-type, set1 Δ, ipl1-2, and set1 Δ ipl1-2 cells. Suppression of chromosomal ade2 mutations by SUP11 yields white colonies, whereas loss of the mini-chromosome yields red colonies (Hieter et al., 1985). Increased chromosome loss is thus measured as an increase in the appearance of red or sectored colonies.
As expected, chromosome loss occurred at very low frequencies (4/1000) in wild-type cells but was elevated more than 10-fold (70/1000) in ipl1-2 cells, even at 25°C (Figure 1C). Deletion of SET1 alone did not affect chromosome stability (3/1000), but it significantly suppressed chromosome loss in ipl1-2 mutant cells (12/1000 or 5/1000; two independent isolates shown in Figure 1C). These data indicate that Set1 normally antagonizes Ipl1 functions in chromosome segregation.
Deletion of SET1 Is Lethal in the Presence of glc7 Mutations
To determine if deletion of SET1 affects phosphorylation events that are regulated by the Glc7 phosphatase, we attempted to delete SET1 in haploid cells harboring the glc7-127 allele, which selectively affects nuclear functions of Glc7, including the dephosphorylation of Ipl1 substrates (Bloecher and Tatchell, 1999; Hsu et al., 2000). However, we never recovered glc7-127 set1 Δ double mutants. To determine if this combination of mutations is lethal, we disrupted SET1 in glc7 cells or isogenic wild-type cells that carried an exogenous, wild-type copy of GLC7 on a URA3 marked plasmid. After confirming the deletion of SET1, we plated cells on media containing 5-FOA to select colonies that had evicted the URA3-GLC7 plasmid. However, glc7-127 set1 Δ double mutants were unable to grow in the absence of the wild-type, plasmid-borne GLC7 gene, suggesting that combination of set1 Δ and glc7 mutations is lethal (Figure 2B).
Figure 2.

- Schematic representation of the plasmid shuffle approach used to combine the glc7-127 mutation or the sds22-6 mutation with SET1 deletion. A URA3-marked plasmid bearing the wild-type GLC7 gene was introduced into either glc7-127 or sds22-6 cells. The SET1 gene was then disrupted. Growth of the resulting cells was compared on rich media (YPD) or on 5-FOA-containing media, which selects for cells that have lost the URA3-marked GLC7 plasmid.
- Serial dilutions (10-fold) of the indicated strains reveals that deletion of SET1 is synthetic lethal with glc7-127 in the absence of the exogenous wild-type GLC7 allele on 5-FOA-containing media (at 25°C).
- Yeast strains with the indicated genotypes were serially diluted 10-fold, spotted onto YPD medium or 5-FOA medium and then grown at indicated temperatures for 3 days. Note the enhanced temperature sensitivity of the sds22-6 set1 Δ cells on 5-FOA-containing media at 30°C and on rich media at 33°C relative to either sds22-6 or set1 Δ single mutants.
- Yeast strains with the indicated genotype were again serially diluted 10-fold, spotted onto YPD medium, and then grown at indicated temperatures for 2 days. As expected, the glc7-127 mutation suppresses the ipl1-2 temperature-sensitive phenotype at 32.5°C. Importantly, ipl1-2 glc7-127 set1 Δ triple-mutant cells are viable, and the temperature-sensitive phenotype of ipl1-2 is also suppressed in these cells.
Sds22 is an essential, nuclear Glc7 binding protein (Hong et al., 2000; Peggie et al., 2002). Temperature-sensitive sds22 mutants exhibit a high rate of chromosome loss and partially suppress the temperature sensitivity of ipl1-2. To further confirm that Set1 is influencing or cooperating with the nuclear functions of Glc7, we deleted SET1 in cells bearing a point mutation in SDS22. Again, we were not able to directly recover sds22-6 set1 Δ double mutants, so we deleted SET1 in sds22-6 mutant cells that carried an extra copy of GLC7 on a URA3-marked plasmid as above, which suppresses the temperature-sensitive sds22-6 mutant phenotype (Figure 2C, bottom panel). Although we defined multiple isolates of sds22-6 set1 Δ cells in the presence of the URA3-GLC7 plasmid, these double mutants exhibited enhanced temperature sensitivity at 30°C and 33°C upon loss of the plasmid (in the presence of 5-FOA; top panel, Figure 2C).
Collectively, our data indicate that Set1 opposes Ipl1 functions or enhances the functions of Glc7-Sds22. In either case, these findings predict that the ipl1-2 mutation should rescue the synthetic lethality of glc7-127 set1 Δ double mutants. Indeed, ipl1-2 glc7-127 set1 Δ triple-mutant cells are viable, and they exhibit significant improvement of growth at 32.5°C relative to ipl1-2 single mutants (Figure 2D). Thus, the lethality of glc7-127 set1 Δ cells is due to an alteration in the balance of Ipl1 and Glc7 functions upon loss of Set1.
Deletion of Other COMPASS Components Suppress ipl1-2
To determine whether the effects of Set1 on Ipl1 function are mediated within the context of the COMPASS complex, we asked whether deletion of four other COMPASS components also suppresses the temperature sensitivity of ipl1-2 cells. Deletion of BRE2, SWD1, and SDC1 (but not SPP1) suppressed the ipl1-2 phenotype at 30°C (data not shown) and 32.5°C (Figure 3A) but could not rescue the lethality of ipl1-2 at higher temperatures (data not shown). Since ipl1-2 suppression is observed upon mutation of three of the four COMPASS subunits tested, Set1 likely acts within COMPASS to modulate Ipl1 or Glc7 functions.
Figure 3.

- (A, B, and C) Yeast strains with the indicated genotypes were serially diluted 10-fold, spotted onto YPD medium, and grown at the indicated temperatures for 2 days.
- (D) Summary of the effects of mutations in genes encoding COMPASS components, PAF1, or the H2B ubiquitylation site on the ipl1-2 phenotype (this work) and H3 K4 methylation (summarized from Krogan et al., [2002], Krogan et al. [2003], and Sun and Allis [2002]).
Set1 Functions in Mitosis Are Independent of Its Role in Transcription Initiation and Early Elongation
Set1 may directly participate in the switch from transcription initiation to elongation, since it associates with a specific phosphorylated isoform of the C-terminal domain of the largest subunit of RNA pol II via the Paf1 elongation complex (Briggs et al., 2001; Krogan et al., 2002; Tenney and Shilatifard, 2005). The genetic effects observed above could reflect alterations in expression of IPL1, GLC7, or another gene important for normal chromosome segregation upon loss of Set1. However, no significant differences in IPL1, ipl1-2, or GLC7 RNA levels were detected in set1 Δ cells relative to wild-type cells (see Figures S1A and S1B in the Supplemental Data available with this article online). Ipl1 protein levels were also unchanged upon loss of SET1 (Figure S1C). Collectively, these data indicate that loss of Set1 does not affect expression of either IPL1 or GLC7.
If the suppression of ipl1-2 by set1 Δ reflects some undefined alteration in gene expression, then deletion of PAF1 should have a similar effect, as this mutation eliminates Set1 association with RNA pol II (Tenney and Shilatifard, 2005). However, ipl1-2 paf1 Δ double mutants exhibit temperature sensitivity and growth defects worse than those of the ipl1-2 single mutants (Figure 3A). Paf1 also stabilizes recruitment of Rad6 to promoters and 5′ coding regions for the ubiquitylation of H2B at K123 (Tenney and Shilatifard, 2005). Mutation of H2B K123 to arginine (R) abrogates COMPASS recruitment and H3 K4 dimethylation (Sun and Allis, 2002) but does not suppress the temperature sensitivity of ipl1-2 at 32.5°C (Figure 3B). Importantly, the lack of suppression of ipl1-2 by paf1 Δ or the H2B K123R mutation demonstrates that ipl1-2 suppression upon deletion of SET1 is independent of Set1 functions in transcription initiation and early elongation.
Deletion of SET1 Does Not Influence H3 S10 Phosphorylation
Although deletion of SET1 does not affect the expression of IPL1 or GLC7, it might affect the activity of these enzymes in vivo. Phosphorylation of H3 S10 is decreased in ipl1-2 mutant cells and increased in glc7 mutant cells (Hsu et al., 2000; Figure S2A). H3 S10 phosphorylation is not changed upon deletion of SET1 and is not recovered in set1 Δ ipl1-2 double mutants (Figure S2A). Therefore, Set1 does not influence global levels of S10 phosphorylation in H3 by Ipl1.
Suppression of ipl1-2 Is Not Linked to Dimethylation of H3 K4
Although deletion of PAF1, mutation of H2B K123, and deletion of SET1 all greatly diminish H3 K4 dimethylation (Krogan et al., 2002), only set1 Δ suppresses the ipl1-2 mutation (Figure 3). Also, there is no correlation between the effects of individual COMPASS mutations on global levels of H3 dimethylation (Krogan et al., 2002) and suppression of ipl1-2 (Figure 3D). These observations suggest that dimethylation of K4 in H3 is not related to modulation of Ipl1 functions by Set1 and COMPASS. To determine more directly whether H3 K4 is involved in either the suppression of ipl1-2 by set1 Δ or the synthetic lethality caused by combination of glc7-127 and set1 Δ, we combined an H3 K4R mutation with the ipl1-2 or glc7-127 alleles. A plasmid bearing the H3 K4R mutation was introduced to cells in which both chromosomal copies of H3 and H4 genes were disrupted, such that the plasmid-borne genes provide the only source of these histones (Zhang et al., 1998). We then compared the growth and temperature sensitivity of cells bearing wild-type H3 or the K4R mutation in the presence or absence of the glc7-127 or ipl1-2 alleles.
Combination of the H3 K4R mutation with the glc7-127 mutation is not lethal (Figure 3C). The survival of these double-mutant cells demonstrates that loss of H3 K4 methylation is not the cause of the lethality observed above in set1 Δ glc7-127 double mutants.
The H3 K4R mutation did suppress ipl1-2 (Figure 3C), allowing improvement of growth at 32.5°C. However, this suppression was noticeably (10- to 100-fold) weaker than that observed in isogenic set1 Δ ipl1-2 cells. Interestingly, an even weaker suppression was observed in triple mutant H3 K4R ipl1-2 set1 Δ cells at 32.5°C (Figure S3). These results suggest that loss of H3 K4 may antagonize, rather than mimic, the effects of Set1 loss on Ipl1 functions. Alternatively, the weak growth of the triple mutants at 32.5°C might reflect loss of several unrelated functions of Ipl1, Set1, and H3 K4.
Collectively, the viability of the H3 K4R glc7-127 cells, the weak suppression of ipl1-2 by the H3 K4R mutation, and the lack of a correlation between the effects mutations in different COMPASS subunits on H3 K4 methylation and ipl1-2 suppression suggest that methylation of substrate(s) other than H3 by Set1 influences Glc7 and Ipl1 functions at the kinetochore.
Conserved Lysines in Dam1 Are Linked to Ipl1 Functions
Dam1 is a prime candidate for a nonhistone substrate of Set1 since previous studies revealed that multiple mutations in Dam1 phosphorylation sites give rise to phenotypes similar to those of the ipl1-2 mutation (Cheeseman et al., 2002a). Importantly, immunoblots with a Dam1-specific antibody reveal that the overall level of Dam1 protein is not altered in set1 Δ, ipl1-2, or double-mutant cells (Figure S2B).
Like other SET domain methyltransferases (Briggs et al., 2001; Jenuwein, 2001), Set1 likely methylates only lysine residues. If Set1 methylates one or more lysines in Dam1, then loss of that methylation site(s) should cause a phenotype similar to the phenotype caused by loss of the enzyme. Therefore, we converted each of the several conserved lysine residues in Dam1 (at positions 7, 129, 132, 138, 194, 233, 252, 256, and 330; Figure 4) separately to alanine (A) and examined the resulting cells for growth defects. We also determined whether any of these K to A mutations suppressed ipl1-2.
Figure 4.

Alignment of Dam1 Protein Sequences in Budding Yeast
Alignment of DAM1 sequences from the indicated yeast are taken from Cliften et al. (2003) and Kellis et al. (2003) as displayed on the Saccharomyces Genome Database website (http://www.yeastgenome.org/). Conserved lysine residues at positions 7, 129, 132, 138, 194, 233, 252, 256, and 330 in DAM1 that were converted to alanine are highlighted, as is the peptide used for antibody production.
Strikingly, only two Dam1 lysine mutations exhibited defective growth, K194A and K233A (Figure 5), and only these same two mutations suppressed ipl1-2. The dam1 K194A allele was temperature sensitive and inviable at 37°C (Figure 5A). However, this mutation suppressed the temperature sensitivity of ipl1-2 cells at 30°C, mimicking the effects of SET1 deletion on the ipl1-2 phenotype. No significant enhancement of suppression was observed upon combination of the dam1 K194A mutation with deletion of SET1 in ipl1-2 cells, indicating that suppression by both mutations occurs through a common pathway (Figure S4).
Figure 5.

- Plate spot assays (as described in previous figures) were used to compare the growth of the indicated strains. Three independent dam1 K194A colonies and three independent ipl1-2 dam1 K194A colonies are shown for comparison.
- Eight independent tetrads from sporulation of dam1 K233A/DAM1 heterozygotes (upper panel). In each case, only two spores were recovered. DNA sequencing revealed that these carry the wild-type DAM1 allele (data not shown), indicating that the dam1 K233A mutation is lethal. However, plate spot growth assays (lower panel) reveal that the lethality of dam1 K233A is suppressed by ipl1-2 and also that the temperature sensitivity of ipl1-2 is suppressed by dam1 K233A.
- Immunoblot of immunoprecipitates from cells expressing native or HA-tagged Dam1 with either native or myc-tagged Set1. Immunoprecipitation was performed with the anti-HA antisera and the immunoblot was probed with either an anti-myc antibody or an anti HA-antibody, as indicated. *myc-Set1.
- (Upper panel) The specificity of the anti-dimethyl K233 Dam1 antisera was confirmed using a dot blot of synthetic Dam1 peptides identical in sequence but containing lysine, dimethyl-lysine, or trimethyl-lysine at the position of K233. Ponceau S staining of a sister blot is shown to confirm equal loading of the peptides. (Lower panel) Wild-type (WT) or K233R mutant HA-tagged Dam1 was immunoprecipitated from wild-type or set1 Δ cells using the anti-HA antibody. An immunoblot of the immunoprecipitates was probed with either anti-HA or the anti-dimethyl K233 Dam1 antisera, as indicated. Immunoprecipitates from a wild-type strain lacking HA-Dam1 is shown as a negative control.
An even more severe phenotype was observed for the K233A dam1 mutation, which is lethal. Sporulation of diploid cells heterozygous for the K233A allele yielded only two viable spores per tetrad (Figure 5B, upper panel), and DNA sequencing confirmed that these live cells carried wild-type DAM1 (data not shown). Strikingly, the lethality of the dam1 K233A mutation is rescued in the presence of the ipl1-2 allele (Figure 5B, lower panel). Moreover, growth of ipl1-2 cells carrying the dam1 K233A mutation is improved at 30°C to an extent similar to that observed upon deletion of SET1. The cross-suppression of the dam1 K233A and ipl1-2 mutants demonstrates not only that the K233A mutant Dam1 protein is expressed but also that K233 has strong functional connections with Ipl1. Combination of both the dam1 K233A mutation with deletion of SET1 in ipl1-2 cells did not further suppress the ipl1-2 phenotype at 32.5°C, indicating that the DAM1 and SET1 mutations affect Ipl1 functions through a common pathway. Moreover, mutation of Dam1 K233 to R or Q is not lethal (data not shown), and the K233Q DAM1 mutation also suppresses ipl1-2 (data not shown).
Dam1 Is Methylated In Vivo at K233
We focused additional studies on Dam1 K233 since the K233A mutation had a more severe phenotype than the K194A mutation. If Set1 methylates K233 in Dam1, then Dam1 should interact physically with Set1 and methylation of Dam1 should be altered in set1 Δ cells. Immuno-precipitation of HA-tagged Dam1 from cells that also expressed a myc-tagged allele of Set1 (Schramke et al., 2001) confirmed that these proteins interact in vivo (Figure 5C). To determine if K233 is methylated in vivo, we raised an antibody to a peptide corresponding to amino acids 222–236 in Dam1 (Figure 4) containing dimethyl lysine at the position of K233. Peptide dot blots confirm that this antiserum is highly specific for the dimethylated Dam1 peptide (Figure 5D, upper panel), although some reactivity was also observed toward a trimethylated peptide. We used this antibody to probe blots of HA-Dam1 immunoprecipitated from wild-type, set1 Δ, or dam1 K233R mutant cells (Figure 5D, lower panel). HA-Dam1 from wild-type cells was recognized well by the anti-dimethyl K233 antiserum, in contrast to minimal or no signal obtained with HA-Dam1 immunoprecipitated from set1 Δ cells or the dam1 K233R mutant. These data confirm that Dam1 is methylated in vivo at K233 and that this methylation is Set1 dependent.
Functional Connections between Dam1 Methylation and Phosphorylation
Dam1 K233 is flanked by conserved serine residues at positions 232, 234, and 235 (Figure 4). Although these serines were not identified previously as targets of Ipl1-mediated phosphorylation, the sequence 232-SKSSQ-236 in Dam1 roughly resembles a consensus sequence ([R/K] × [TS] [ILV]) identified for Ipl1 targets (Cheeseman et al., 2002a). Mutation of these sites reduces the number of slow-migrating Dam1 phosphoisoforms detected by immunoblot (Cheeseman et al., 2002a; Figure 6A), suggesting these sites are phosphorylated in vivo. Mass spectrometric analyses of peptides generated upon cleavage of immunopurified HA-tagged Dam1 were also consistent with phosphorylation at one or more of these serines (Figure S5). In addition, a synthetic peptide corresponding to the sequence NIGMS KSSQGHV in Dam1 (amino acids 228–239) is phosphor-ylated by a recombinant GST-Ipl1 fusion protein in vitro (Figure 6B). However, Ipl1-mediated phosphorylation is diminished significantly in a sister peptide containing dimethyl-lysine at the position of K233. Trimethyl-lysine at this position also inhibited phosphorylation by Ipl1 (data not shown).
Figure 6.
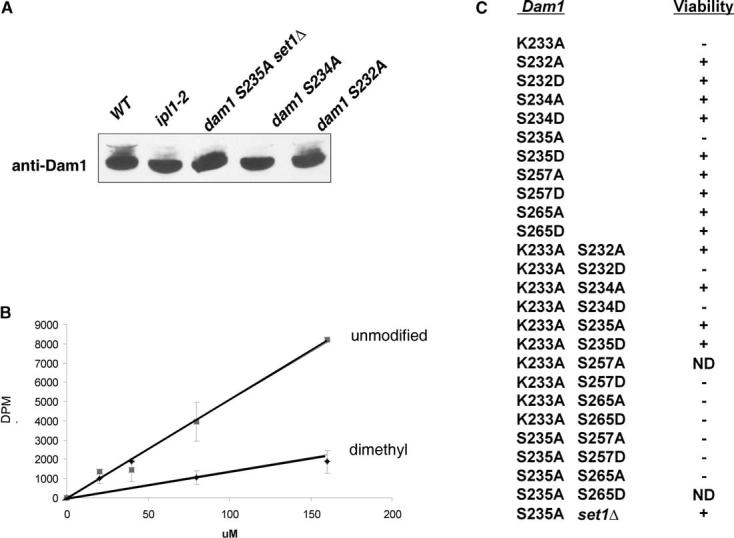
- Immunoblot analysis reveals diminishment of slower migrating Dam1 phosphoisoforms (Cheeseman et al., 2002a) upon mutation of IPL1 or mutation of S232, S234, or S235 in Dam1.
- Kinase assays demonstrate that a recombinant GST-Ipl1 fusion protein phosphorylates a peptide corresponding to Dam1 amino acids 228–239, indicating that one or more serines flanking K233 are recognized by Ipl1 as substrate in vitro. Shown are the averages of triplicate assays at each amount of peptide used. Error bars represent the standard deviations. Phosphorylation is inhibited by the presence of dimethyl-lysine at the position of K233.
- Summary of the effects of the indicated dam1 mutations on cell viability. Note the suppression of the lethality of the K233A mutation by either the S232A or S234A mutations and the cosuppression of the K233A and S235A lethality in K233A S235A double mutants. The S235A lethality is also suppressed in set1 Δ cells. ND: not determined due either to a failure of mutant cells to sporulate (K233A S257A) or failure to recover heterozygous DAM1/dam1 cells (S235A S265D).
To determine if the genetic interactions between Dam1 K233 and Ipl1 observed above might reflect changes in the phosphorylation of these flanking serines in vivo, we combined the K233A mutation with substitution mutations at these sites. We mutated each serine to alanine to mimic the nonphosphorylated state and to aspartate (D) to mimic the phosphorylated state.
Individual mutations at S232 or S234 in DAM1 to either A or D had no effect on cell growth or viability (Figure 6C). Strikingly, the S232A and S234A mutations rescued the lethality of the K233A mutation, but the S232D and S234D mutations did not. Mutation of two serines previously identified as Ipl1 targets, S257 and S265 (Cheeseman et al., 2002a), to A or D also did not rescue lethality of the K233A mutation. These data suggest that loss of K233 (or loss of methylation at this site) is specifically compensated by neutral charges provided by the flanking S232A or S234A mutations. These results are also consistent with the suppression of K233A by ipl1-2 observed above and suggest that K233 methylation might normally be required to limit phosphorylation of these sites by Ipl1.
Interestingly, the S235A DAM1 mutation is lethal, whereas the S235D mutation is not, indicating that phosphorylation of S235 may be essential for Dam1 functions (Figure 6C). S235A is the first single-serine mutation in Dam1 to cause a lethal phenotype. Remarkably, the S235A allele is viable in combination with the K233A mutation. These two lethal mutations suppress one another. The S235D allele also rescues the lethality of the K233A mutation, indicating that the charge of the residue at position 235 is not important if K233 is not present. Mutation of more distal Ipl1 target sites (S257 or S265; Cheeseman et al., 2002a) in Dam1 to D or A does not rescue S235A lethality, indicating the cassette of modifications from S232 to S235 are uniquely linked in function.
The above data suggest that loss of negative charge at S235 and likely loss of phosphorylation is lethal only when K233 is present and available for methylation. Indeed, the lethality of the S235A mutation in DAM1 is also suppressed by deletion of SET1 (Figure 6C), further demonstrating a connection between S235 functions and methylation. Collectively, the suppression of the lethality of K233A by ipl1-2 and the suppression of S235A by set1 Δ, together with the genetic interactions between the K233A mutation and the S232A, S234A, and S235A mutations and our in vitro kinase assays, indicate that levels of Ipl1 phosphorylation of S232, S234, or S235 are fine-tuned by Set1 methylation at K233.
Discussion
Our data reveal unexpected functional connections between the Set1 methyltransferase and phosphorylation events governed by the Ipl1 kinase and the Glc7 phosphatase. Loss of Set1 suppresses chromosome segregation defects caused by the ipl1-2 allele and is synthetic lethal with the glc7-127 allele. The mitotic functions of Set1 require Bre2, Swd1, and Sdc1, indicating that Set1 functions in the context of the COMPASS complex to modulate Ipl1-Glc7 functions in chromosome segregation.
Previous studies have revealed a role for Set1 and COMPASS in gene transcription that requires Paf1 and ubiquitylation of H2B at K123 (Briggs et al., 2001; Krogan et al., 2002; Tenney and Shilatifard, 2005). However, our data demonstrate that deletion of PAF1 or mutation of H2B K123 cannot suppress ipl1-2. Therefore, the suppression of ipl1-2 upon deletion of SET1 is independent of COMPASS functions in transcription initiation and early elongation.
Prior to our studies, H3 K4 was the only known substrate of Set1. However, loss of H3 K4 methylation is not likely the molecular basis for the genetic interactions between SET1, IPL1, and GLC7 that we observe. First, mutations in SET1, PAF1, or H2B K123 all globally diminish H3 K4 methylation (Tenney and Shilatifard, 2005), yet only SET1 deletion suppresses ipl1-2. Second, we found no correlation between the effects of deletion of other COMPASS components on H3 K4 methylation (Krogan et al., 2002) and suppression of ipl1-2 (Figure 3). Third, mutation of H3 K4 to R suppresses ipl1-2 more weakly than does deletion or mutation of SET1, and the H3 K4R mutation is not synthetic lethal with glc7-127. Finally, chromatin immunoprecipitation results (T. Tripic, M. Coombes, and S.Y.R.D., unpublished data) indicate that little or no H3 K4 methylation occurs at centromeres in S. cerevisiae, consistent with the replacement of H3 with Cse4 in centromeric nucleosomes (Meluh et al., 1998; Westermann et al., 2003).
Unlike centromeres in S. pombe and most other organisms (Nakayama et al., 2001; Noma et al., 2001), centromeres in S. cerevisiae are not flanked by heterochromatic repeat elements (Cleveland et al., 2003), and this yeast does not contain HP1-like proteins or Suv39 methyltransferases. H3 K9 is not methylated in S. cerevisiae (Rea et al., 2000), and mutations in H3 S10 do not affect chromosome segregation (Hsu et al., 2000). Moreover, we find no evidence of global changes in phosphorylation of S10 in the absence of Set1. Therefore, the effects of Set1 loss on Ipl1 functions that we observe do not likely reflect indirect effects on modifications at S10 or K9 in H3. Rather, our results indicate that these effects are mediated through Set1-mediated methylation of at least one nonhistone substrate, Dam1.
How might Dam1 methylation at K233 or K194 contribute to proper chromosome segregation? By analogy to the effects of histone methylation on the occurrence of other posttranslational modifications of the histones (Zhang and Reinberg, 2001), K233 methylation might directly affect phosphorylation of neighboring serines. This model is consistent with our observation that Ipl1-mediated phosphorylation of methylated Dam1 peptides is inhibited in vitro (Figure 6B), as well as our genetic data that reveal functional connections between K233 and S232, S234, and S235 (Figure 7). Our data indicate that prevention of K233 methylation by set1 Δ allows improved phosphorylation of Dam1 by the crippled ipl1-2 kinase, as reflected by suppression of the ipl1-2 phenotype, but might allow too much or too persistent phosphorylation by wild-type Ipl1. Conversely, the suppression of the lethality of the DAM1 K233A allele by flanking S to A mutations or by the ipl1-2 mutation (but not by S to D mutations) strongly suggests that negative effects associated with loss of Dam1 methylation can be countered by decreased phosphorylation at these sites (Figure 7). Several previous findings that indicate a balance in the phosphorylation and dephosphorylation of IPL1 and GLC7 substrates is essential for normal cell growth and chromosome segregation (Francisco and Chan, 1994; Francisco et al., 1994). Our results strongly suggest that the region between K194 and S235 is a critical module in Dam1 that is regulated by both phosphorylation and methylation.
Figure 7.
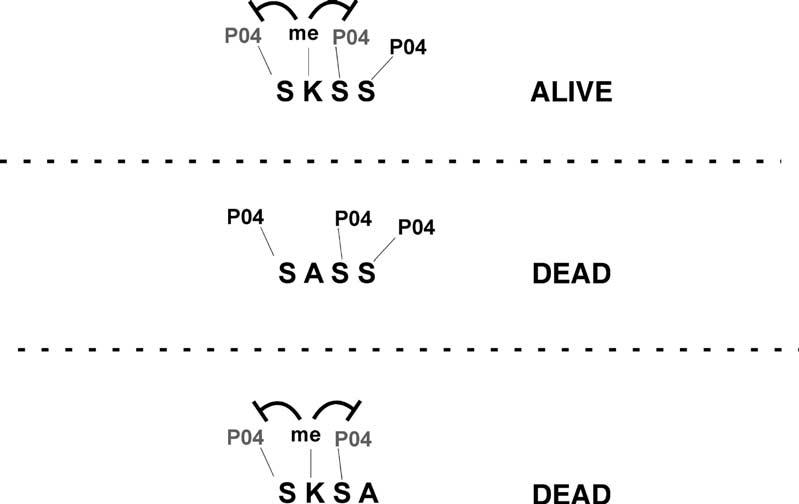
Model for Modulation of Dam1 Phosphorylation by Methylation of K233
Deletion of SET1 suppresses chromosome segregation defects associated with the ipl1-2 mutation, indicating that Set1 normally opposes the functions of Ipl1. Our data suggest that this effect is mediated at least in part by methylation of Dam1 by Set1. We propose that methylation of K233 negatively regulates phosphorylation of the two flanking series, S232 and S234. Phosphorylation of S235 may or may not be influenced directly by Set1. When K233 is converted to alanine, preventing methylation by Set1, phosphorylation of S232 or S234 may increase to a level that interferes with Dam1 functions and kinetochore integrity. Conversely, if K233 is intact and available for methylation, Dam1 may become hypophosphorylated when S235 is mutated to alanine, leading to loss of viability.
Cycles of phosphorylation and dephosphorylation of Dam1 and other kinetochore proteins allow resolution of improper monopolar spindle attachments and reformation of proper bipolar attachments (Cheeseman et al., 2002b; Shang et al., 2003). These phosphorylation events may control protein-protein interactions as well as interactions between kinetochore components and microtubules. Lysine methylation in other settings also influences interactions between proteins. Chromodomain proteins recognize K9 in H3, for example (Fischle et al., 2003; Nielsen et al., 2002), and methylation of K4 in H3 limits association of repressor complexes with certain promoters (Zegerman et al., 2002). Methylation of Dam1 and/or other kinetochore proteins might influence kinetochore assembly, stability, or even constitute part of the signal that inhibits continued cycles of kinetochore dissolution once proper microtubule attachments are made. Another intriguing possibility is that Dam1 methylation might provide a “memory” function to distinguish kinetochores that have previously passed through mitosis from newly synthesized kinetochores, similar to the suggested function of histone methylation as a “memory” of gene transcription (Ng et al., 2003; Turner, 2002).
Interestingly, deletion of SET1 is not lethal, but mutation of K233 to A in Dam1 causes cell death. Mutation of K194 in Dam1 also causes a temperature-sensitive phenotype that is not observed in set1 Δ cells. These differences in phenotype suggest either that K194 and K233 in Dam1 play roles in limiting Ipl1 functions that are independent of methylation or that these residues are subject to modification by additional enzymes. K233 and K194 might be methylated by another methyltransferase or even subject to acetylation or ubiquitylation.
Deletion of SET1 was reported previously to suppress mutations in several DNA-repair checkpoint genes, and Set1 interacts physically with Mec3 (Corda et al., 1999). Loss of Set1 causes hyperphosphorylation of the replication protein Rfa2, which is dependent on the checkpoint-signaling kinase Rad53 (Schramke et al., 2001). Together with our results, these findings suggest that Set1 may influence the functions of multiple kinases involved in several distinct cellular processes.
Aurora kinases are often deregulated in cancer cells and tumors that exhibit aneuploidy and loss of genome integrity (Katayama et al., 2003). Mutations in histone methyltransferase genes are also associated with cellular transformation and cancer formation (Schneider et al., 2002). The MLL gene, which is similar to Set1, is often a target of translocation in mixed lineage leukemias (Tenney and Shilatifard, 2005). Our studies in yeast raise the possibility that Aurora kinases and methyltransferase activities may crossregulate each other in mammalian cells. If so, then changes in the balance of these enzyme activities may contribute both to tumor suppression and to oncogenesis.
Experimental Procedures
Strains and Growth Conditions
Yeast strains used in this study are listed in Table S1, and details of strain constructions are provided in the Supplemental Experimental Procedures. Yeast were propagated according to standard procedures either in rich media (YPD) or in appropriate selective media (SC).
Plasmid Construction and Mutagenesis
The entire coding sequence of DAM1 was cloned into BamHI and XhoI sites of pRS406 (Sikorski and Hieter, 1989). Site-directed mutations in DAM1 were constructed using Quickchange protocols (Stratagene) and the DAM1-pRS406 plasmid.
The glc7-127 allele was also cloned into pRS406 using a PCR fragment templated with genomic DNA extracted from the KT1640 strain. The wild-type GLC7 plasmid used in the experiments shown in Figures 2A and 2B was described previously (Wu and Tatchell, 2001).
Full length SET1 was cloned into NotI and XhoI sites of pRS406 to generate SET1-pRS406. G951S-pRS406 was also constructed using Quickchange protocols (Stratagene) and the plasmid SET1-pRS406.
Peptide Synthesis and Generation of Dimethyl-K233 Dam 1-Specific Antisera
A peptide corresponding to amino acids 222–236 in Dam 1 containing dimethyllysine at position K233 was synthesized by Genemed, Inc. and then used to raise polyclonal rabbit antisera and to affinity purify the antisera. Additional peptides corresponding to amino acids 222–240 or 228–239 in Dam1 containing unmodified, dimethyl- or trimethyllysine at position K233 were synthesized by the Peptide Synthesis Core Facility of the M.D. Anderson Cancer Center.
Immunoprecipitation of Dam1 and Immunoblot Using Anti-Dimethyl Dam1 K233 Serum
Cell extracts were prepared from 500 ml of cells grown in YPD to an OD600 of 0.8. Cells were collected by centrifugation, washed two times with water, and resuspended in 5 ml lysis buffer (50 mM Tris 8.0, 150 mM Nacl, 1% NP40) with proteinase inhibitors. The resus-pension was flash frozen in liquid nitrogen and ground into powders in a coffee mill with dry ice. After thawing at 4°C, cellular debris was pelleted by centrifugation at 5000 × g for 15 min. The supernatant was then preclarified by incubating with100 μl protein A agarose (Upstate Biotech) at 4°C for 1 hr. HA-Dam1 was then immunoprecipitated using 200 μl HA affinity matrix (Roche) at 4°C for 4 hr. The HA affinity matrix was collected and washed three times with lysis buffer and then resuspended in 100 μl lysis buffer with SDS-PAGE loading dye. Proteins were resolved by 12% SDS-PAGE, transferred to Immun-Blot PVDF membrane (Bio-Rad), blocked for 1 hr with Tris-buffered saline/0.05% Tween 20 containing 5% nonfat milk, followed by 1 hr incubation with anti-dimethyl Dam1 K233 serum at a final dilution of 1/5000. After incubation with anti-rabbit horseradish peroxidase-conjugated secondary antibodies (Amersham Biosicence), bands were detected using the Pierce Supersignal Westpico Chemiluminescence system.
Protein Kinase Assays
Kinase assays were conducted as described previously (Li et al., 2002) using an S6 kinase assay kit procedure (Upstate Biotechnology).
Supplementary Material
Acknowledgments
The authors thank D. Drubin, C. Shang, S. Anderson, S. Tanaka, J. Workman, V. Geli, and C.D. Allis for sharing advice, results, and reagents. We thank Tamara Tripic and Madelene Coombes for performing chromatin immunoprecipitation experiments described in the Discussion, and Wei Qi and Cherie Coco for technical assistance with several experiments. We thank Waleed Nasser for technical assistance with the mass spectrometric analyses of Dam1. We thank D. Edmondson for help in the analysis of H3 phosphorylation at S10. We especially thank J. Davie for much expert advice. DNA sequencing, peptide synthesis, and proteomics analyses were performed by UTMDACC core facilities funded by NCI core grant CA16672. Mass spectrometric analyses were also supported in part by a grant from the Goodwin Foundation to R.K. K.Z. is a Rosalie B. Hite Fellow, and J.A.L. is supported by NIH T32 HD07325. This research was supported by a grant from the Robert A. Welch Foundation to S.Y.R.D. (G1371) and a grant from the NIH to J.M.S. (R01GM62181).
Footnotes
Supplemental Data
Supplemental Data include five figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://www.cell.com/cgi/content/full/122/5/723/DC1/.
References
- Bloecher A, Tatchell K. Defects in Saccharomyces cerevisiae protein phosphatase type I activate the spindle/kinetochore checkpoint. Genes Dev. 1999;13:517–522. doi: 10.1101/gad.13.5.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buvelot S, Tatsutani SY, Vermaak D, Biggins S. The budding yeast Ipl1/Aurora protein kinase regulates mitotic spindle disassembly. J. Cell Biol. 2003;160:329–339. doi: 10.1083/jcb.200209018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Botstein D. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics. 1993;135:677–691. doi: 10.1093/genetics/135.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR, 3rd, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 2002a;111:163–172. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Drubin DG, Barnes G. Simple centromere, complex kinetochore: linking spindle microtubules and centromeric DNA in budding yeast. J. Cell Biol. 2002b;157:199–203. doi: 10.1083/jcb.200201052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: From epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Corda Y, Schramke V, Longhese MP, Smokvina T, Paciotti V, Brevet V, Gilson E, Geli V. Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat. Genet. 1999;21:204–208. doi: 10.1038/5991. [DOI] [PubMed] [Google Scholar]
- Courtwright AM, He X. Dam1 is the right one: Phosphoregulation of kinetochore biorientation. Dev. Cell. 2002;3:610–611. doi: 10.1016/s1534-5807(02)00332-5. [DOI] [PubMed] [Google Scholar]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducat D, Zheng Y. Aurora kinases in spindle assembly and chromosome segregation. Exp. Cell Res. 2004;301:60–67. doi: 10.1016/j.yexcr.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Edmondson DG, Davie JK, Zhou J, Mirnikjoo B, Tatchell K, Dent SY. Site-specific loss of acetylation upon phosphorylation of histone H3. J. Biol. Chem. 2002;277:29496–29502. doi: 10.1074/jbc.M200651200. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco L, Chan CS. Regulation of yeast chromosome segregation by Ipl1 protein kinase and type 1 protein phosphatase. Cell. Mol. Biol. Res. 1994;40:207–213. [PubMed] [Google Scholar]
- Francisco L, Wang W, Chan CS. Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol. Cell. Biol. 1994;14:4731–4740. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieter P, Mann C, Snyder M, Davis RW. Mitotic stability of yeast chromosomes: A colony color assay that measures nondisjunction and chromosome loss. Cell. 1985;40:381–392. doi: 10.1016/0092-8674(85)90152-7. [DOI] [PubMed] [Google Scholar]
- Hong G, Trumbly RJ, Reimann EM, Schlender KK. Sds22p is a subunit of a stable isolatable form of protein phosphatase 1 (Glc7p) from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 2000;376:288–298. doi: 10.1006/abbi.2000.1715. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22:451–464. doi: 10.1023/a:1023789416385. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 2002;277:10753–10755. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell. 2003;11:721–729. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Bachant J, Alcasabas AA, Wang Y, Qin J, Elledge SJ. The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev. 2002;16:183–197. doi: 10.1101/gad.959402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM. Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell. 1998;94:607–613. doi: 10.1016/s0092-8674(00)81602-5. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr. Opin. Genet. Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. USA. 2002;99:90–94. doi: 10.1073/pnas.221596698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- Noma K, Grewal SI. Histone H3 lysine 4 methylation is mediated by Set1 and promotes maintenance of active chromatin states in fission yeast. Proc. Natl. Acad. Sci. USA. 2002;99(Suppl 4):16438–16445. doi: 10.1073/pnas.182436399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Allis CD, Grewal SI. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science. 2001;293:1150–1155. doi: 10.1126/science.1064150. [DOI] [PubMed] [Google Scholar]
- Peggie MW, MacKelvie SH, Bloecher A, Knatko EV, Tatchell K, Stark MJ. Essential functions of Sds22p in chromosome stability and nuclear localization of PP1. J. Cell Sci. 2002;115:195–206. doi: 10.1242/jcs.115.1.195. [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Sassoon I, Severin FF, Andrews PD, Taba MR, Kaplan KB, Ashford AJ, Stark MJ, Sorger PK, Hyman AA. Regulation of Saccharomyces cerevisiae kinetochores by the type 1 phosphatase Glc7p. Genes Dev. 1999;13:545–555. doi: 10.1101/gad.13.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem. Sci. 2002;27:396–402. doi: 10.1016/s0968-0004(02)02141-2. [DOI] [PubMed] [Google Scholar]
- Schramke V, Neecke H, Brevet V, Corda Y, Lucchini G, Longhese MP, Gilson E, Geli V. The set1Delta mutation unveils a novel signaling pathway relayed by the Rad53-dependent hyperphosphorylation of replication protein A that leads to transcriptional activation of repair genes. Genes Dev. 2001;15:1845–1858. doi: 10.1101/gad.193901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang C, Hazbun TR, Cheeseman IM, Aranda J, Fields S, Drubin DG, Barnes G. Kinetochore protein interactions and their regulation by the Aurora kinase Ipl1p. Mol. Biol. Cell. 2003;14:3342–3355. doi: 10.1091/mbc.E02-11-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- Tenney K, Shilatifard A. A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: Linking leukemogensis to covalent modifications of chromatin. J. Cell. Biochem. 2005;95:429–436. doi: 10.1002/jcb.20421. [DOI] [PubMed] [Google Scholar]
- Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 1999;97:99–109. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- Westermann S, Cheeseman IM, Anderson S, Yates JR, 3rd, Drubin DG, Barnes G. Architecture of the budding yeast kinetochore reveals a conserved molecular core. J. Cell Biol. 2003;163:215–222. doi: 10.1083/jcb.200305100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Avila-Sakar A, Wang HW, Niederstrasser H, Wong J, Drubin DG, Nogales E, Barnes G. Formation of a dynamic kinetochore-microtubule interface through assembly of the Dam1 ring complex. Mol. Cell. 2005;17:277–290. doi: 10.1016/j.molcel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Wu X, Tatchell K. Mutations in yeast protein phosphatase type 1 that affect targeting subunit binding. Biochemistry. 2001;40:7410–7420. doi: 10.1021/bi002796k. [DOI] [PubMed] [Google Scholar]
- Zegerman P, Canas B, Pappin D, Kouzarides T. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J. Biol. Chem. 2002;277:11621–11624. doi: 10.1074/jbc.C200045200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 1998;17:3155–3167. doi: 10.1093/emboj/17.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Reinberg D. Transcription regulation by his-tone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.