
Figure 1.
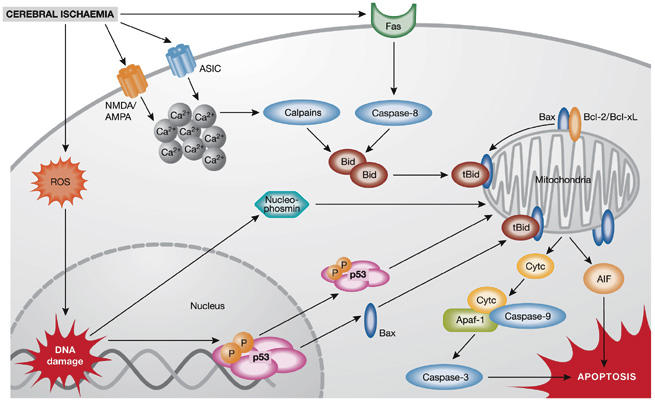
Mechanisms of ischaemic neuronal cell death. Ischaemic neuronal death is mediated by a plethora of intracellular mechanisms, such as increased intracellular calcium levels, reactive oxygen species (ROS), DNA damage and mitochondrial dysfunction. Post-ischaemic cytotoxic calcium influx is mediated through the stimulation of N-methyl-D-aspartate (NMDA), D,L-α-amino-3-hydroxy-5-methyl-isoxazolpropionic acid (AMPA) glutamate receptors or acid-sensing ion channels (ASICs). Increased intracellular calcium, ROS accumulation and activation of Fas death receptors result in activation of caspase-8 or calpains and mediate cleavage of Bid to truncated Bid (tBid), which integrates the different death pathways at the mitochondrial checkpoint of apoptosis. At the mitochondrial membrane tBid interacts with Bax, which is usually neutralized by anti-apoptotic B-cell leukaemia/lymphoma 2 (Bcl-2) family proteins Bcl-2 or Bcl-xL. Dimers of tBid and Bax form pores in the outer mitochondrial membrane, thereby releasing cytochrome c (Cytc) or apoptosis-inducing factor (AIF), which execute caspase-dependent or caspase-independent cell death, respectively. After release into the cytosol, cytochrome c complexes with apoptotic protein-activating factor-1 (Apaf-1) and procaspase-9 to form the apoptosome, which further activates executor caspases, such as caspase-3. By contrast, AIF translocates rapidly to the nucleus where it mediates large-scale DNA fragmentation and cell death in a caspase-independent manner. In addition, nuclear pathways of neuronal cell death are activated in response to DNA damage, for example, through phosphorylation (P) and activation of p53 or mitochondrial translocation of nucleophosmin.